Deep learning in omics: a survey and guideline
- 杂志: Briefings in functional genomics
- IF: 3.133
- 分区: 2区
Introduction
1943年MCP原型首次提出【1】--> Rosenblatt提出感知机模型【2】--> 1974年首次提出了back propagation(bp,反向传播算法)【3】--> 2006年Hinton首次解决了梯度消失问题并显示了DL技术的重大潜力【4】。之后,因为:
- 数据量大,数据维度高且复杂使得传统Machine Learning(ML)方法变得吃力;
- 硬件发展使得计算能力达到要求;
- DL社区,特别是想Google的大公司推动了DL技术的发展;
DL得到了空前的发展。其中也包括一系列bioinformatics领域的应用:
- 利用电子病历数据进行疾病预测【5,6】;
- 生物医学图像分类【7-10】;
- 生物信号处理【11-13】;
现代omics研究面临两个难题:
- 在诊断、预测等应用领域,实验室方法昂贵且费时;
- 现存数据复杂、多态,传统方法已经难以处理;
但这些难题都有望被深度学习克服。
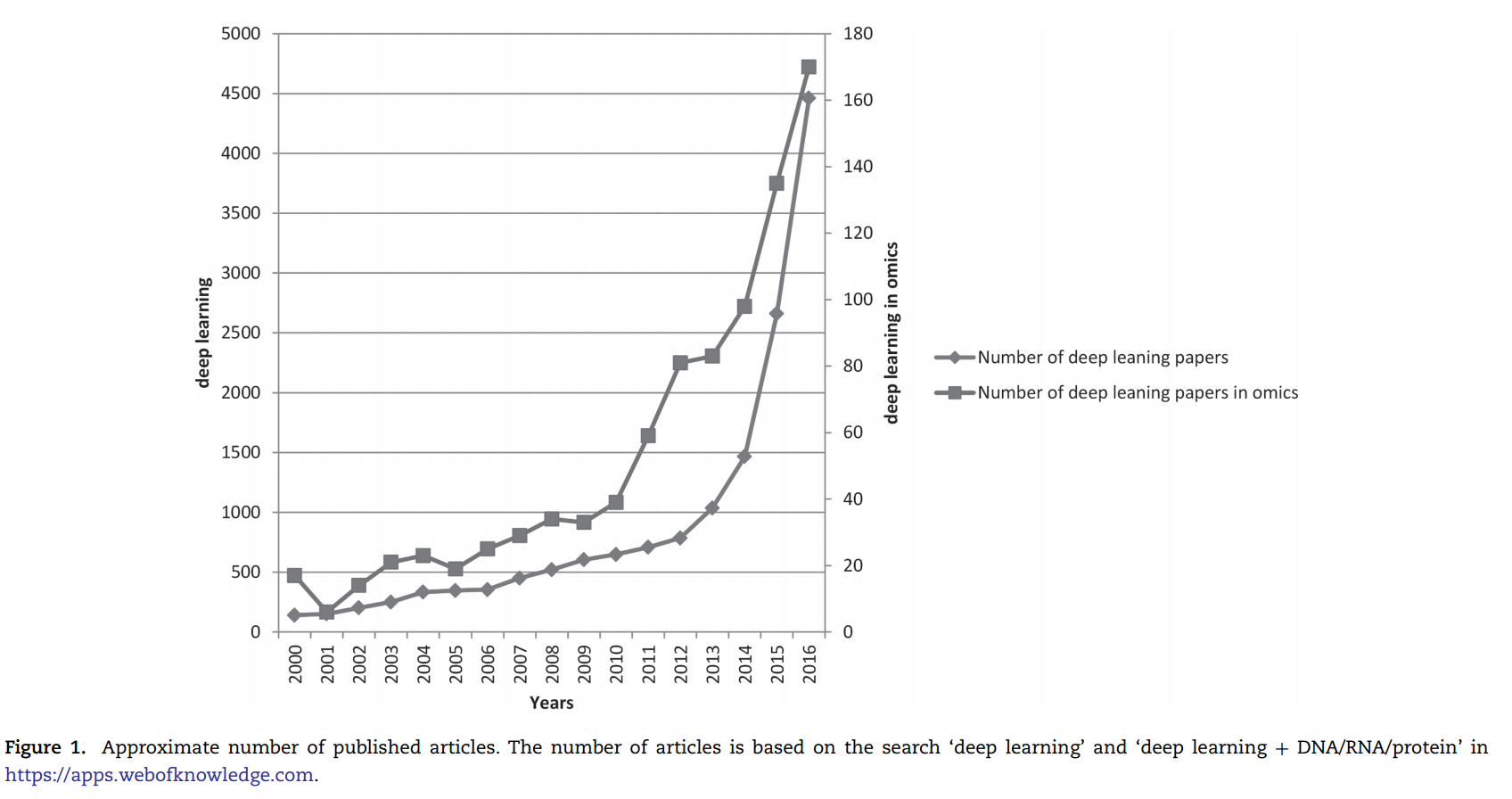
DL Models in Omics
这里涉及到很多基础的DL知识的,就不赘述了。
DNNs
这里指代了MLP、AEs、RBMs和DBNs【4】等多种模型。其中DBNs解决了蛋白质残基连接预测【27】和RNA结合蛋白位点预测问题【28】。
CNNs
略。
RNNs
略。
Applications in omics
Genomics
预测DNA序列的功能单元,如复制域(replication domain)、转录因子结合位点(transcription factor binding sites, TFBS)、转录起始点、启动子、增强子、基因删除位点等。
复制域:DNA复制时间调控的单位。 转录因子:指能够结合在某基因上游特异核苷酸序列上的蛋白质,这些蛋白质能调控其基因的转录。方法是转录因子可以调控核糖核酸聚合酶(RNA聚合酶,或叫RNA合成酶)与DNA模板的结合。 启动子:指一段能使特定基因进行转录的脱氧核糖核酸(DNA)序列。启动子可以被RNA聚合酶辨认,并开始转录合成RNA。在核糖核酸(RNA)合成中,启动子可以和调控基因转录的转录因子产生相互作用,控制基因表达(转录)的起始时间和表达的程度,包含核心启动子区域和调控区域,就像“开关”,决定基因的活动,继而控制细胞开始生产哪一种蛋白质。 增强子:是DNA上一小段可与蛋白质(反式作用因子;transacting factor,也就是转录因子)结合的区域,与蛋白质结合之后,基因的转录作用将会加强。增强子可能位于基因上游,也可能位于下游。且不一定接近所要作用的基因,甚至不一定与基因位于同一染色体。这是因为染色质的缠绕结构,使序列上相隔很远的位置也有机会相互接触。
- 2015年,将预训练、DNN和hidden Markov model结合的模型实现了对复制域分类的突破【34】;
- 2016年,【35】通过使用convolutional/highway的MLP框架,依据TFBS来对基因序列进行分类,AUC达到了0.946;
- 【36】使用CNN模型来对启动子进行分子,实现了0.9左右的ACC;
- 【37】使用3层全连接来进行增强子和启动子的分类,达到了93.59的ACC;
- 【38】开发了一个浅层CNN模型-CNNdel,来进行基因删除位点的预测,使用来自1000 Genomes Projects的数据进行训练,也得到了超过其他方法的结果。
总体来说,CNNs逐渐压过了DNNs称为主流,并且开始出现了CNNs+LSTMs的杂交模型。
预测基因表达。比如目标基因的表达量、预测基因功能、建模基因调控网络等。
- 【39】利用GEO的microarray数据,训练了一个DNN model来预测目标基因的表达量,明显好于Logistic;
- 【40】基于stAEs和MLP,通过基因变异的基因型来预测基因表达;
- 【41】提出了一个基于CNN-biLSTM的模型,称为DanQ,来预测非编码区域的功能,实现了97.6的AUPR;
- 【42】使用RNN来建立基因调控网络,超越了其他的方法;
CNNs依然是此领域的主要模型,而在研究比较少的基因调控网络领域,还是主要以RNN为主。
探索基因和疾病的关系。
- 2017年,【43】使用MLP模型来预测cancer risk和生存,使用的是TCGA的数据,并和cox弹性网得到了类似的表现;
- 【44】使用CNNs来预测序列变异对近端CpG位点(DNA甲基化)的影响,得到了0.854的AUC;
- 【45】,即DeepCpG,在单细胞领域来预测甲基化位点,其使用的是CNN-RNN的杂交模型,并且模型可以解释;
在预测甲基化的领域,还是以CNNs为主,RNNs可能会有所应用,但一般也要CNNs配合。而在探索基因和疾病的关系领域,主要使用DNNs,并且AEs和DBN也有所应用。
Transcriptions
预测RNA序列的结构,比如预测RBP结合位点、可变剪切位点和RNA类型。
- 2015,基于DBN模型即可探索潜在的结合位点【28】,使用到的数据有RNA序列信息、RNA二级结构和RNA三级结构信息,相对于之前的方法MRE下降了22%;
- 【84】开发了一个deep CNNs model来进行剪切点的分类,称为DeeSpline,提高了预测ACC; c.2017年,基于MLP的模型的成功实现了pre-miRNAs和psudo hair-pins的分类,并得到了0.968的ACC【46】;
探索RNA和疾病的关联。
- 2017年,基于DBN的一个分类模型,成功利用miRNA属性进行疾病的分类,平均提高了6%-10%【47】;
- 【48】使用转录组数据并结合DNN模型,来确定各种药物在不同生物系统下的药理学性质,此模型的效果要好于SVM;
Proteomics
蛋白质结构预测。比如蛋白质二级结构预测、蛋白质模型质量评估、protein contact map等。
- 【49】通过st-sp AEs来预测二级结构和扭转角,使用原始氨基酸序列作为输入,达到了82%的ACC;
- 【36】使用DNN替代SVM来进行蛋白质模型质量评价,就PCC从0.85提高到了0.9;
- 【50】结合了两个deep residual networks来预测contacts,其接受sequence conservation information和evoluationary coupling作为输入,得到了最高的F1-score;
蛋白质功能预测。
- 【51】中,使用CNN模型来预测蛋白质功能,使用蛋白质三级结构作为输入,得到了87.6%的准确率;
- 【52】使用LSTM来预测4种类型蛋白质的功能,使用的是原始氨基酸序列作为输入,得到了99%的ACC;
预测蛋白质交互作用、蛋白质亚细胞定位或其他功能。
- 【53】使用stAEs来预测蛋白质交互作用,得到97.19%的ACC;
- 【54】使用CNNs模型来自动检测亚细胞定位,在每个细胞定位分类任务中得到了91%的ACC,在每个蛋白质上得到了99%的ACC;
开源软件
很多人将开发的模型、工具的开源代码放在了网上,或者做成了web服务,现在将其列举如下:
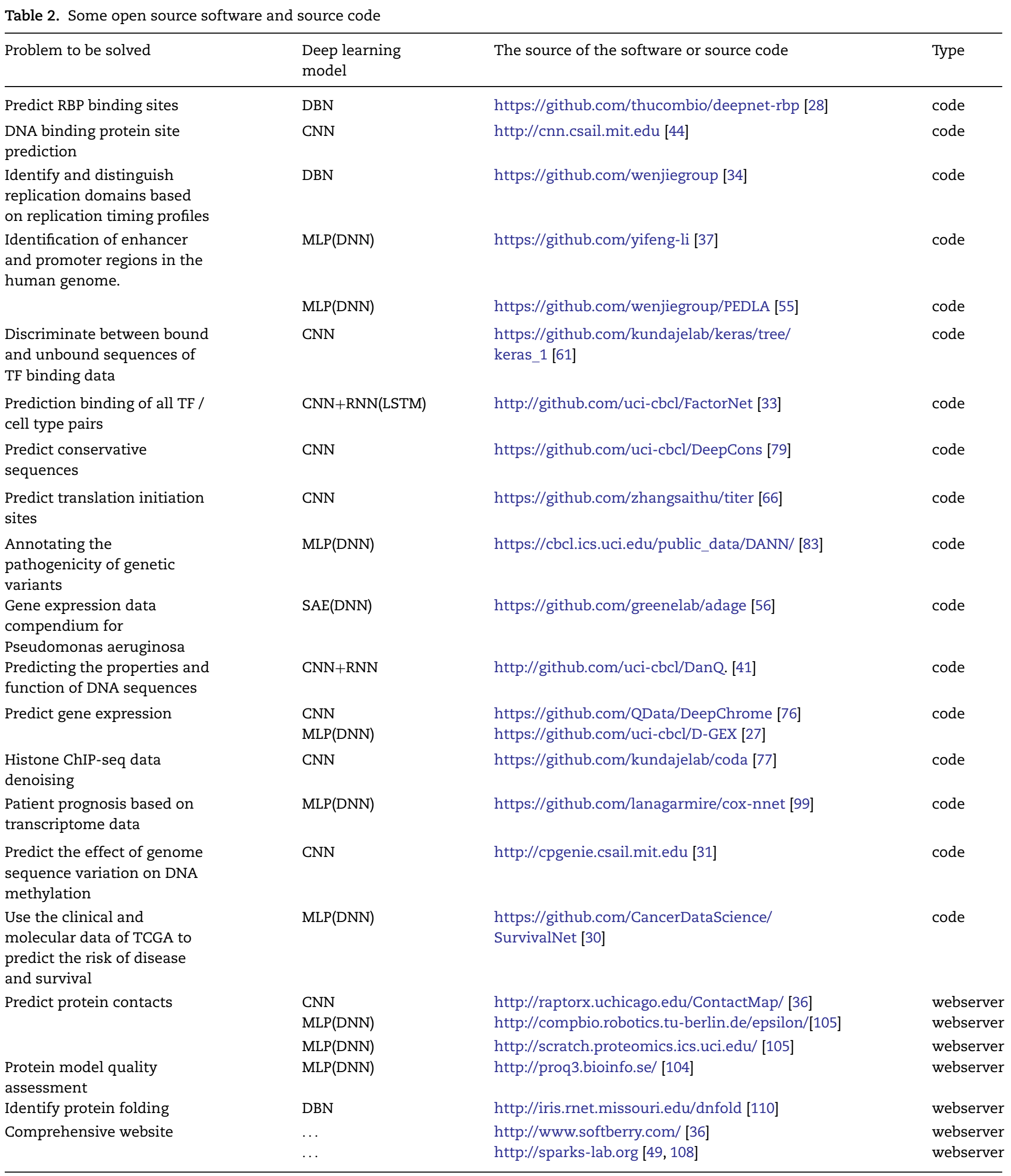
使用DL来解决omics问题
这里总结了使用DL来进行omics研究的流程。
数据获得
以下是常用的omics数据库:
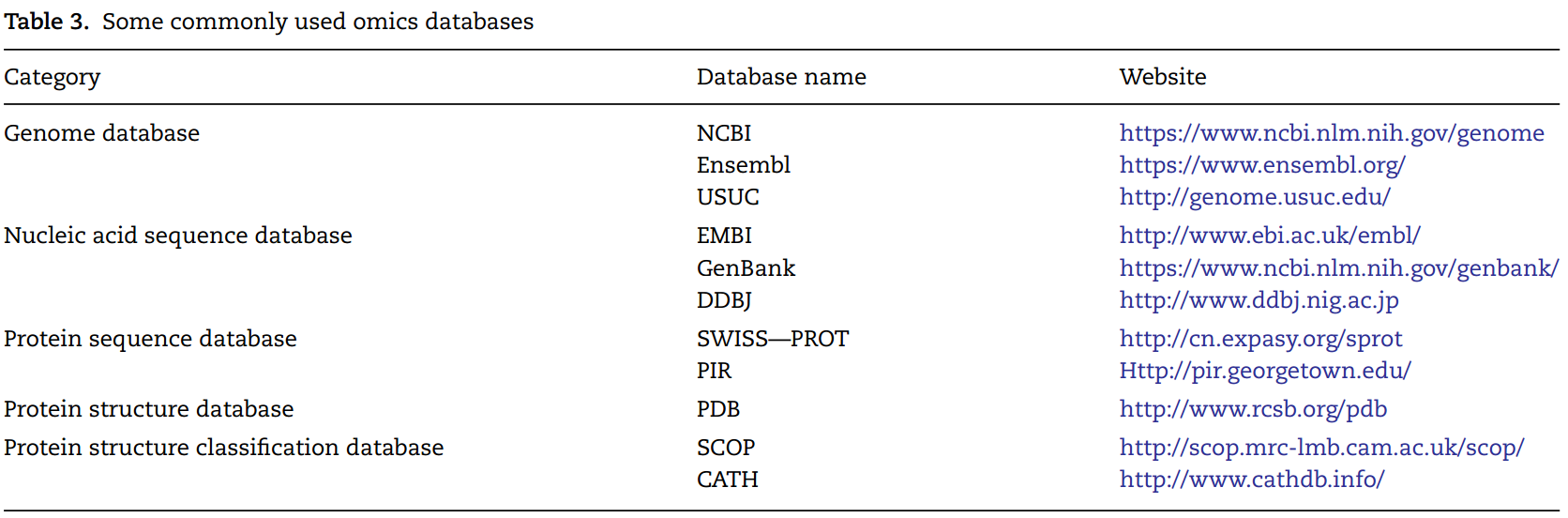
需要对其进行一定的处理。
数据预处理
data cleaning。
- 缺失值填补,异常值处理:可使用k近邻、regression或决策树等方法来进行填补;
- 去除重复的数据:把相似性超过某个阈值的样本删除;
- 处理噪声:使用聚类方法、regression或binning;
这个工作是time-consuming和labor-intensive,也没有什么统一的标准。可以使用一些方便的软件包:
OpenRefine【124】或DataKleenr。Normalization。
主要的作用是将数据缩放至一个合适的范围内。
- 如果我们的数据比较集中,并且不服从正态分布,没有涉及到距离、相关性的计算,我们可以使用min-max normalization。比如图像数据。
- 如果数据大约服从正态分布,并且我们希望保留样本间距离关系,则可以使用zero-mean normalization。比如基因表达谱数据等。这也是最常用的normalization方法。
Dimensionality reduction。
删除一些无关的变量,使用AEs来进行降维,或者使用PCA等等。
编码
one-hot encoding
\(N\)长度的DNA序列被编码为\(4\times N\)的矩阵,蛋白质序列被编码成\(20\times N\)的矩阵,其中的每个值是\(0\)或\(1\)。
position-specific scoring matrix(PSSM, PWM, PSWM)
也可以用来编码核苷酸序列或蛋白质序列,这里使用wiki的例子进行解释:
我们有以下9条序列:
GAGGTAAAC TCCGTAAGT CAGGTTGGA ACAGTCAGT TAGGTCATT TAGGTACTG ATGGTAACT CAGGTATAC TGTGTGAGT AAGGTAAGT首先将每个位置上的频数进行计算,称为PFM(position frequency matrix):

然后每个位置归一化为概率,得到PPM(position probability matrix):

最后将概率转换为权重,即使用\(\log2(M_{k,j}/b_k)\),其中\(b_k=1/|k|\),其中\(k\)表示每个位置上的离散值有多少个,对于氨基酸序列\(k=20\),而对于核苷酸序列则\(k=4\),最后得到PWM(position weight matrix):
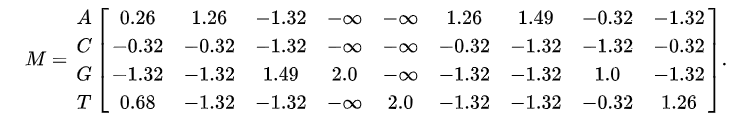
然后使用其来编码数据即可。
有一些现有的程序可以实现(比如PSI-BLAST),在这些算法中,其计算过程更加复杂,但基本思想是一致的。
这样编码是认为在序列的不同位置,相同的标签也应该有不同的编码。比如在序列开始的A和在序列结束时的A应该有不同的值。
如果某个位置上,A出现的次数明显比CGT更多,则其自然在此位置有更高的权重。如果在另外的位置上,A出现的次数比较少,则其就有较小的权重。如果一次都没有出现,权重是\(-\infty\)
另外,权重的大小还和类别数有关。比如在某位置上,氨基酸序列中A(丙氨酸)出现的频率是0.7,在DNA序列中A(腺嘌呤)出现的概率也是0.7,但最终氨基酸序列计算的编码要比DNA序列计算的编码大。这是因为在20分类中得到0.7的概率要比4分类更难,自然应有更高的赋值。
PAM(point accepted mutation)matrix and BLOSUM(blocks substitution)matrix
这两个矩阵是序列的替换记分矩阵,常用于序列比对,即其衡量了序列上的两个值间的相似程度。那么同样的,其也可以用来作为序列的编码。详情请见
除了以上3种方法外,对于蛋白质序列,还有autocovariance method和conjoint triad method 【126】方法。
另外,还有一些方法致力于将多种类型的数据进行整合分析【128,49,50,98,129】。
模型选择
- 尽量针对不同的任务使用不同的架构;
- 可以将多种不同的架构进行结合,从而得到更好的结果;
- 时刻关注最先进模型的研究,关于新技术。
模型训练
- 考虑一下硬件配置是否支持;
- 注意数据集要分成training、validation和testing,本研究作者常用的分发为70%用作training和validation,30%用作testing;
- 合理选择激活函数,本研究作者建议的隐藏层激活函数为ReLU和maxout;
- 考虑试验一下dropout、early stop和weight decay,来预防过拟合。
评价
推荐使用交叉验证。
深度学习框架
这里总结了一下框架,但内容有点过时,就不贴了。
机会和挑战
- 数据量:omics数据量一般不足,并且有imbalance的问题;
- 解决数据量不足:zero-shot learning【135】、one-shot learning【136】和GAN【137】; 解决imbalance:resamling、cost-sensitive learning【138】。
- 数据质量:omics数据一般来自不同平台,数据质量比较难以保证;
- 解决:上面提到的数据清洗过程。
- 计算花销:有硬件门槛;
- “黑盒”问题:无法进行解释;
- 关于“黑盒”问题,也有所进展。比如【139】提出的Deep Motif Dashboard,就为TFBS的分类提供了一种可视化策略。
- 模型选择和训练:选择一个合适的模型和合适的超参数是困难的;
当然,未来,还有一些其他的DL概念会对omics的研究产生影响:
- reinforcement leanring 【140】
- incremental learning 【141】
- transfer learning 【142】
结论
DL技术非常适合解决omics的问题。
Questions
这篇文章的一些结论或者内容感觉有些老了,之前以为是19年的文章,可能会比较新。但读到才发现可能是18年的。
- 但还是有所收获,特别是其介绍的一些序列编码方法是第一次听说。总结了一些文章的github。另外,其提到的文章中也有一些值得关注的。比如那个使用RNN来建网的方法,还有提到的一系列基于DBN的方法。*



