PAMOGK: A Pathway Graph Kernel based Multi-Omics Clustering Approach for Discovering Cancer Patient Subgroups
- 杂志: bioRxiv
- IF: ---
- 分区: ---
- github
Introduction
cancer是一个分子多样性基本,能够分离出不同的分子亚型对于疾病的诊断和治疗将有重大的意义;
多个组学的数据有助于研究者对cancer病人的认识【48,46,9】,因此基于多组学的cluster methods被广泛研究【35】:
最简单的就是把所有组学的数据concatenated,但带来了问题:维度灾难、每个组学的权重相同。
iClusterBayes及其变体【42, 30, 29】、LRACluster应用一些降维方法和正则化方法来试图解决上述问题。
另一种策略:分别在每个组学中进行聚类,然后再将多个组学的结果集成。这一类方法有一致性聚类【31】、PINS【34】、COCA【17】等。其缺点是无法捕捉各个组学间的相互关系。
之后,一批intermediate integration algorithm被开发出来【36】:
- SNF【51】先为每个数据类型构建一个patient similarity network,然后使用一些算法(比如message passing)来将这些network融合在一起;
- 【28】则提出一种降维策略,其维度被转换到多个组学间最大协方差的方向;
- 【27】提出了JIVE;
- MCCA【54,5】则将CCA扩展到了多组学领域;
- 【21,7】将谱聚类【50】扩展到了多组学领域,谱聚类可以看做是一种kernel method;
- 【56,45,13】一系列generic multi-view kernel cluster methods也被扩展到多组学领域。
这里对multi-omics(多组学)和multi-view(多视图)进行一下解释。
multi-view表示的是对于同一种事物,我们使用多种途径或多种角度来进行描述,由此形成的数据称为multi-view datasets,而基于此的机器学习门类称为multi-view learning。
multi-omics则是一个组学、生物信息学的概念,即使用多个组学的数据来共同分析。注意到,这多个组学可能并不是同一样本的数据。
所以对于两者来说,是有交叉的:当multi-omics数据是同一批病人的不同组学数据时,这实际上就是一个multi-view问题。
当然,两者在概念上也有一定的区别,multi-view更多是一个机器学习的概念,而multi-omics是一个生物信息学的概念或者组学的概念,有点类似CV和医学影像处理之间的关系。
还有一个多模态学习(multimodal),这是multi-view的一个子集,指的是人的不同感官的数据,或对于同一个事物使用不同的方法收集到的数据。
可能单纯地使用多个组学还不足以明晰病人间的相似性,配合分子网络的稀疏性【8】可能更好。
本研究提出PAMOGK方法。其将每个病人视为一个有标签的无向图,结合一个novel graph kernel - SmSPK来提取病人的相似性。然后可以将病人分到不同的亚组中,并提供了每个pathway、组学对聚类所做的贡献。
本研究将此方法应用到TCGA-KIRC数据集(somatic mutations、gene expression、protein expression)中,得到了4个亚组,并在生存上存在差异。
Methods
\(S\)表示病人集合,我们的目的是希望得到一个划分\(\{C_i\}\),\(M\)表示pathways的数量,\(D\)表示组学数据数量,\(N\)表示病人的数量。
overview
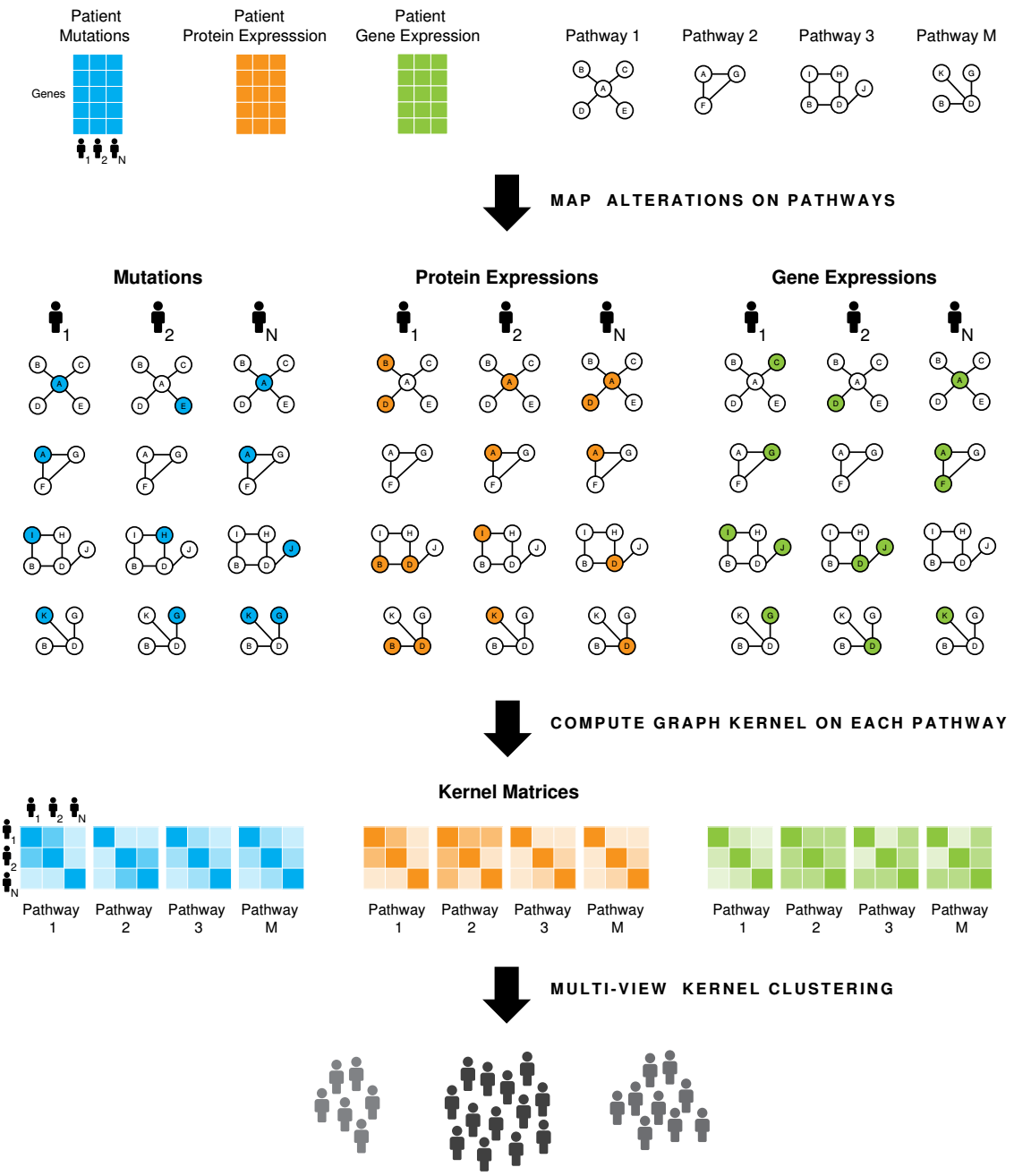
主要包括3步:
- 对于每个组学,将其映射到pathway中,每个patients被表示成\(M\times D\)的graph;
- 使用SmSPK来定量两个patients间的相似性(kernel matrices);
- 使用multi-view clustering algorithms,利用第2步得到的kernel matrices,将patients分离到有意义的亚组中。
step1-Patient graph representation
对于pathway i和patient j,我们得到graph \(G_i^j=(V_i, E_i, l_i^j)\)。其中\(V_i\)是该pathway中的基因set,\(E_i\)是该pathway中存在的基因之间的调控关系(没有方向),\(l_i^j\)表示patient j在各个基因上的值(因为本研究中使用的都是binary的数据,所以这里的值就是0-1)。
显然,对于同一个pathway来说,所有patients的graph的结构是相同的,不同的只是那个\(l_i^j\)。
最终我们得到\(N\times M\times D\)个labeled pathway graphs。
step2-Computing Multi-View Kernels with Graph Kernels
graph kernel function输入两个graphs,输出这两个graphs间的相似性【49】,已经有许多graph kernel function【43,4,33】,但这些都是输入的不同结构的graphs来通过结构的差异来确定两个图的相似性,并不适合当前的任务,所以本研究自己建立了一个新的graph kernel。
基于shortest path graph kernel【4】,开发得到SmSPK:
首先进行label propagation(即随机游走),得到smoothed graph,其计算公式为:
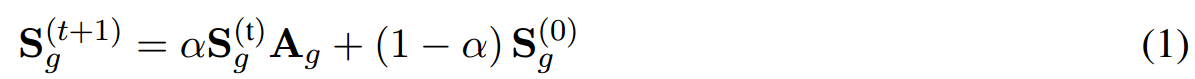
其中,\(g\)表示pathway graph,\(t\)表示step,\(A_g\)是pathway \(g\)的度归一化邻接矩阵,\(S_g(0)\)就是\(l\),\(\alpha\in[0,1]\)是用来定义光滑度的参数。
进行迭代计算,直到收敛,然后我们得到了label smooth graphs。
对于两个人在相同通路下的相似性,这样计算:

其中,\(s_p^{(i)}\)表示smoothed图\(G_g\)在patient \(i\)上的最短路径\(p\)上的节点组成的向量,\(P\)表示最短路径的数量。
经过上述计算,我们得到\(M\times D\)个\(N\times N\)的matrices。
注意,这里的逻辑是,我们将图进行随机游走,如果图上每个节点的值是先验的概率,则经过随机游走后得到的是最终会出现在各个点上的概率。这一定程度上是综合了节点值和图结构的embedding,然后用这个embedding来计算两个图的相似性。
要深入理解这里graph kernel的内容,可能需要去了解相关知识。
这里为了能够将多组学信息融合,本研究了存在的multi-view kernel clustering methods,发现multiple kernel k-means中matrix-induced regularization(MKKM-MR,【25】)效果最好,所以本研究采用这个方法,当然也可以使用其他的方法:
Multiple Kernel K-Means with Matrix-Induced Regularization(MKKM-MR):
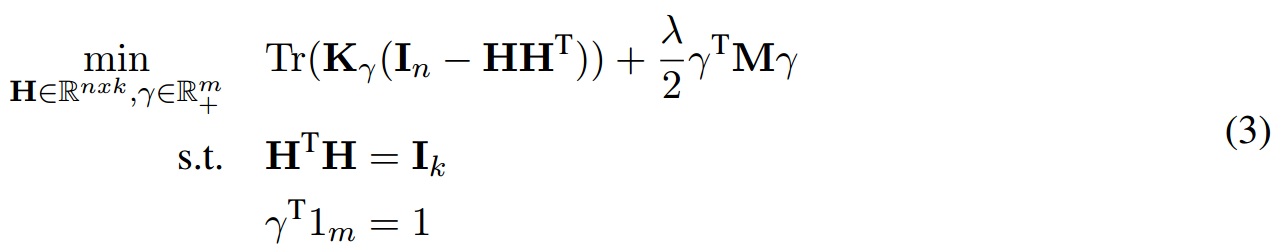
大体的思想是去找一个所有kernel matrices的最佳组合来做聚类。
其中\(m\)表示使用的kernel matrices的数量,\(n\)表示样本的数量,\(k\)表示的聚类数量,\(\gamma=[\gamma_1,\cdots,\gamma_m]\)是在融合所有kernel matrices时每个kernel matrix的权重,\(K_{\gamma}=\gamma_1D_1+\cdots+\gamma_mD_m\),\(D_i\)是第\(i\)个kernel matrix,\(M\)是各个kernel matrices间的关系的度量,\(I_x\)是\(x\times x\)的identity matrix,\(1_m\)是值全为1的m维向量,\(\lambda\)是来调节正则化的参数。
Average Kernel K-Means (AKKM):就是使用Kernel K-means(KKM)【39】,使用的Kernel是所有的Kernel matrices的平均。
Localized multiple kernel K-means(LMKMM)【13】。
Results and Discussion
数据
pathway数据:来自NDEXBio【38】的NCI-PID(2019-04-24下载),专注于癌症研究,将和omics数据中没有任何重叠genes的pathway去掉,还剩下165个pathways。
病人的分子数据和临床数据:
- 使用TCGA PanCancer的KIRC数据(来自Synapse),使用了三种类型的数据(RNAseq gene expression,RPPA,somatic mutation);
- 只使用其中的primary solid tumour samples;
- 删除了在超过一半样本中没有表达的genes;
- 对于RPPA,只使用没有磷酸化的蛋白表达;
- 通过GDC得到临床信息;
- 在3个组学上都有,而且生存信息完整的病人被保留,共得到361个patients,236个right-censored,125个passed away。
将分子数据融入graph中:
- 计算gene和protein的z-scores,然后对于z-score大于1.96的看做是overexpressed,z-score小于-1.96的看做而是underexpressed。
- 最终得到5中binary的数据,即somatic mutation、RNAseq的over/underexpressed data、RPPA的over/underexpressed data。
实验设置
- 得到了825个kernel matrices (165 x 5)。
- 试验了聚类数\(k=2,3,4,5\),12种不同的\(\alpha\),并试验了上面提到了3种multi-view clustering methods。
- 将kernel matrix中非0值少于1%的kernel matrices删除以增加运行速度。
- \(\lambda\)用grid-search选择。
- 评价指标使用生存分析【2,22,1,12】,即绘制K-M曲线和计算log-rank p值来进行比较。
新的graph kernel的必要性
必要性:其他的graph kernel只能检测拓扑结构的相似性,而对于拥有相同拓扑结构但不同值的graph,其检测能力不足,所以开发了SmSPK。
比较:使用the densest kernels数据(overexpressed genes)来作为评价数据集,然后比较了SmSPK、shortest path kernel【4】、propagation kernel【33】、Weisfeiller Lehman subtree kernel【43】。
软件:Grakel library。
计算:将每一个kernel matrix中的值都分到不同的bins中,然后计算这个kernel matrix在不同bins中的频率,然后将所有kernel matrices的频率进行平均,结果如fig2a显示:
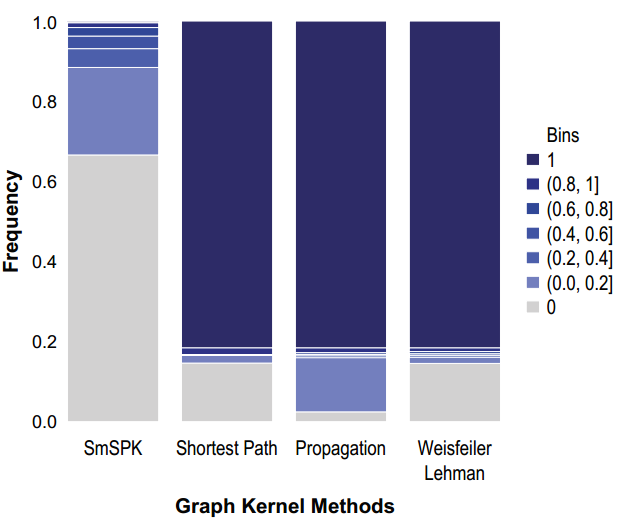
结果:可以看到,除了SmSPK之外,其他的方法大多认为patients间的相似性应该是1,这说明结构相同的graphs,这些kernel无法检测出其之间的差别,若SmSPK对这些差别是敏感的。
应该使用MKKM-MR
这里比较了多种multi-view clustering methods,每种聚类方法在使用的时候可以自动进行超参数的调整(聚类数\(k\),光滑参数\(\alpha\),RBF kernel的参数\(\gamma\),MKKM-MR的\(\lambda\))。
并且也使用了不同的kernel methods,防止kernel methods对multi-view clustering methods的影响。但因为之前已经证明了除了SmSPK之外的methods无法找到patients之间的差异性,所以这里只选择了shortest path kernel(最相近的),另外还选择了RBF kernel来看graph kernel是否有必要。结果如fig2b所示:
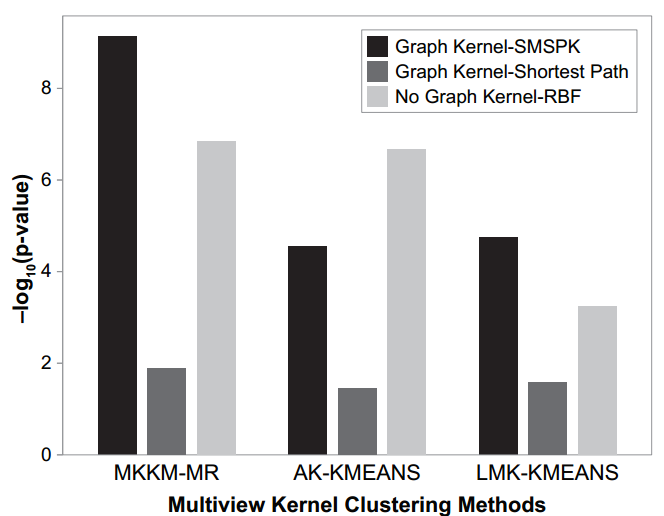
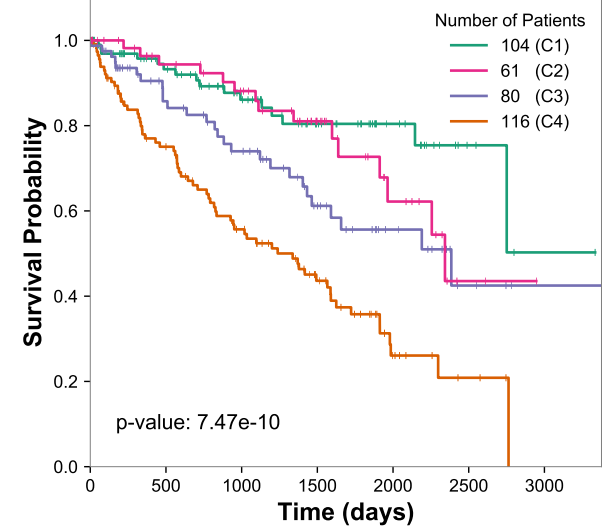
结果显示,MKKM-MR配合SmSPK,在\(k=4,\alpha=0.3,\lambda=8\)时,得到最好的结果(p=7.4e-10),其K-M plots在下面(\(k=3\)时也不错,在附录中有展示):
和当前最好的多组学方法比较
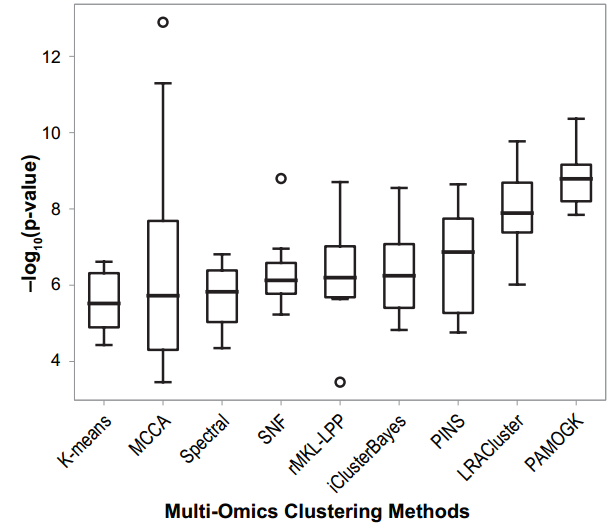
和其中8种方法进行了比较,这些方法在【36】中有介绍,这里使用的超参数是各自在benchmark study中的配置(除了聚类数),聚类数则是设置一个最大值,然后选择各自最好的。
抽取了300个patients,进行聚类。将上述过程重复10次,来比较不同方法。fig3b显示PAMOGK的效果是最好的。
另外,还比较了一下运行速度:

PAMOGK的kIRC亚组分析
和临床指标的关系:发现和年龄、性别是没关系的,但和其他的都有关系,结果如下:

cluster 1(预后最好的一组)的病人有更低的stage和grade,而cluster 4(预后最不好的一组)的病人有更高的stage和grade,这表明PAMOGK的亚组是有临床意义的。
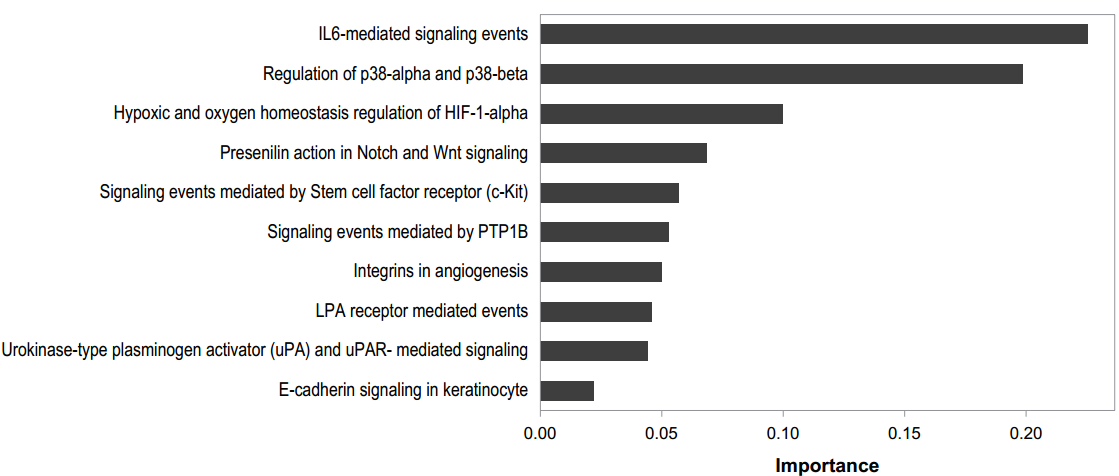
也可以通过\(\gamma\)来判断哪个组学、哪条通路是更加重要的:
IL-6 mediated signaling events + gene overexpression是最重要的;
通过平均权重,可以得知gene expression是最重要的data type,而protein expression几乎没有影响(可能是因为RPPA数据中的genes数量太少了);
最高权重的通路是IL-6 mediated signaling events。
Conclusion
本研究的局限性:使用的TCGA数据,即bulk数据,这些数据是多种类型细胞的混合,有比较强的intra-tumor heterogeneity【10】。所以未来可以将PAMOGK应用到单细胞数据上。
还有许多方向可以探索:将binary扩展到continuous;SmSPK是基于shortest path kernel而来,可能有其他更好的选择;考虑graph的方向性;考虑蛋白质交互作用网络。
Questions
这篇论文写的还是挺不错的,很清楚。但其中关于multi-view clustering的技术和graph kernel的技术我并不太擅长,这可能影响了我对本文的理解,所以之后要多看一些这方便的内容了。
本研究中比较让我感兴趣的是其中的fig2a,这里显示其改进得到的SmSPK kernel确实可以比其他的kernel更加关注于graph labels的内容,而不把宝都压在graph topology。我对原本的graph kernel的方法并不太清楚,所以我也不知道作者的那一部分修改使得效果这么立竿见影。但随之而来一个要问的问题是:SmSPK是否能够在graph topology不同的graphs中起到效果,即它是否既能够有效的捕获labels information,又能够提取topology的information呢?
本研究的最后表示,使用bulk的数据已经是一个缺点了(:(),看来单细胞是大势呀。
关于kernel的方法可以赶紧学一下,说不定能够和深度学习产生一些火花。



