Integrated Multi-omics Analysis Using Variational Autoencoders: Application to Pan-cancer Classification
- 杂志: arXiv
- IF: --
- 分区: --
- github
写在前面的话:
可以说本文件为我生动的上了一课--数据备份的重要性。为了测试自己编写的python-gui-bolg管理系统,却没有将当前写完的笔记进行备份,尽管慎之又慎,但还是覆盖了几篇我辛辛苦苦写下的文件,本篇就是其中之一。尽管尝试了多种数据恢复软件,但依然无法从被覆盖的文件中找到其以前的版本,所以我放弃了挣扎,只能重新再写一遍。
为此,我进行的补救措施有:首先查看了所有我写过的十几篇笔记,查看有哪些有问题;将当前写笔记的文件环境同步到坚果云,利用其可以历史备份的功能;重新编写了我的博客管理系统的备份功能,现在其备份不会被覆盖,而是每次备份时都会备份称为单独的一个文件夹;为hexo博客系统的source文件夹添加git管理,并在每次generate的时候都commit一次。
以后再要测试一些新的东西的时候,一定要把其可能影响到的文件保护好,一旦丢失(比如先复制一份到其他文件夹下),想要找回实在是太难了(一晚上没有睡,就整这个了。。)
Introduction
- 组学数据,或者更进一步的多组学数据,存在“维度灾难”的问题,需要使用变量筛选或降维的方法来进行预处理。
- 基于DL领域的特征提取方法(如VAE)在其他领域已经显现出其作用,但在组学领域还没有得到足够的研究。
- 本文提出了OmiVAE模型,其可以:
- 基于VAE的降维;
- 整合classification到VAE模型中进行end-to-end的学习,可以得到task-oriented的features;
相关工作
受到CV和NLP领域的启发,DL领域的许多算法被应用到多组学领域:
- 【6】使用AE来对多组学数据进行降维,并在此基础上使用kNN和SVM得到了有生存差异的亚组。
- 【7】提出了基于LSTM的VAE模型用于建模代谢组学、蛋白质组学的时间序列数据。
以上提到的研究构建的模型都不是end-to-end,
- 【8】提出了提出了一个end-to-end的multi-omics模型,使用factorization AE来预测无病生存期,得到了0.664(膀胱癌)和0.746(胶质瘤)的avPR。
至于癌症分类领域:
【9】将PCA和spAE结合来学习gene expression的表示来进行癌症分类。
【10】则使用sdAE。
【11】使用VAE来提取gene expression的latent representation,然后再分析其和表型间的联系。
【12,13】使用VAE来分析methylation数据。
【14】将GCN和relation network结合,来进行乳腺癌亚型分类,并使用到了PPI数据。
这篇看过
【15】开发了一个CNS系统来进行癌症分类,其基于DNA methylation数据和RF。
以上的模型大多集中特定的某个癌症类型,至于在pan-cancer领域:
- 【16】使用knn模型和遗传算法来对TCGA Pan-cancer数据进行处理,得到了95.6%的预测acc。
- 【17】将gene expression转变成2D images,然后使用CNN进行33类分类任务,得到了95.59%的avACC。
- 【18】使用CNN应用到相同的数据上,得到了95.7%的ACC和95%的ACC(34类分类)。
Methods
数据
TCGA pan-cancer的RNAseq数据和DNA methylation数据,共有33种癌症:
- gene expression (RNAseq):11538 samples,其中741 normal samples,外显子标记60483个,使用的数据格式是FPKM的log2转换值。
- DNA methylation:使用450K的数据,共有485578个探针,9736个样本,其中746个normal samples,使用的数据格式是beta值(探针对应CpG位点的甲基化率)。
数据预处理
gene expression:
- 将位于Y染色体的基因(594)、全部都是0值的基因(1904)和有超过10%样本缺失的基因(248)去除;
- 最终去除2440个分子特征,保留58043。
DNA methylation:
- 将无法映射到hg38标记(89512)、在Y染色体上的(346)、超过10%样本是缺失值的(414)probes去掉;
- 最终得到392761个CpG位点,然后将这些CpG位点根据其所属染色体,分到23个组中,每个组平均拥有的probes数量是17077。
缺失值使用均值填补,FPKM的log2值被归一化到0-1间,Beta值不用变(因为作为率,其本身就在0-1之间),然后保留在两类数据集中都有的样本(9081,其中407个normal samples)。
网络架构
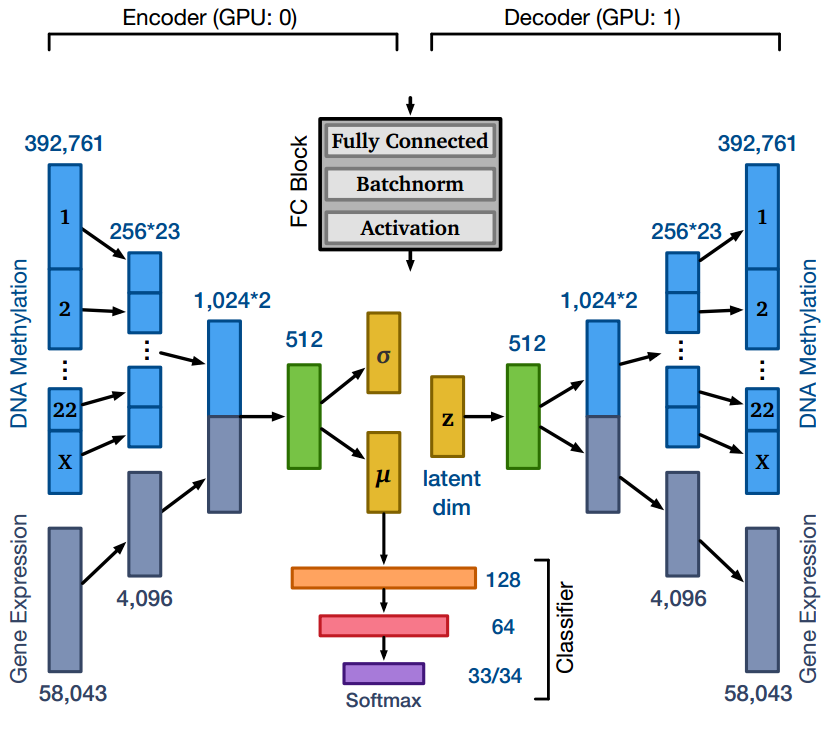
如果fig1所示:
关于VAE原理的部分不再赘述,只是将其ELOB公式粘贴在下面,供参考。
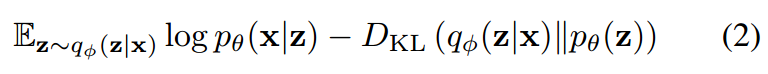
关于网络架构的要点:
为了能够处理methylation data的高维度,先分别对单个染色体上的位点进行映射(每个染色体映射至256),然后再合并。
将合并后的methylation特征再映射一次(1024),才和也映射到1024的expression特征(-->4096-->1024)cat。
瓶颈层是128维/2维(用于可视化)。
从瓶颈层的均值部分接一个(-->128-->64-->33/34)的网络进行end-to-end的分类训练,所以使用的是下面的loss:
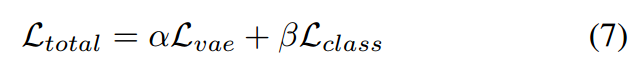
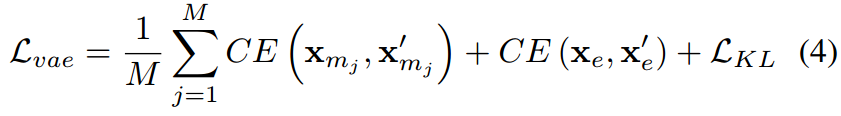
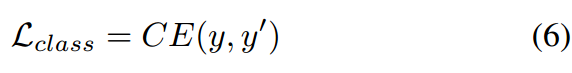
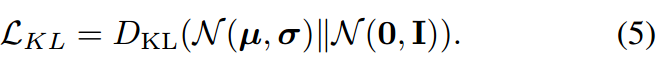
这里有两个training phase:
- 如果是unsupervised phase,就使用式4(实际上就是ELBO的具体形式,或者是式7,但beta值设为0)来训练VAE部分;
- 如果是supervised phase,则先进行unsupervised phase(看做是预训练),然后使用式7(其中beta值是1)进行微调。
Implementation:
- 使用的隐层激活函数是ReLU,decoder的输出层是sigmoid,分类使用softmax。
- 使用PyTorch(1.1)。
- 使用了2块1080Ti,一个用来放decoder,一个用来放encoder训练。
- Adam,lr=10-3,batch size=32,early stopping。
Results
使用10-CV来评价性能
无监督过程
先把瓶颈层的维度设置成2,看VAE的可视化能力:
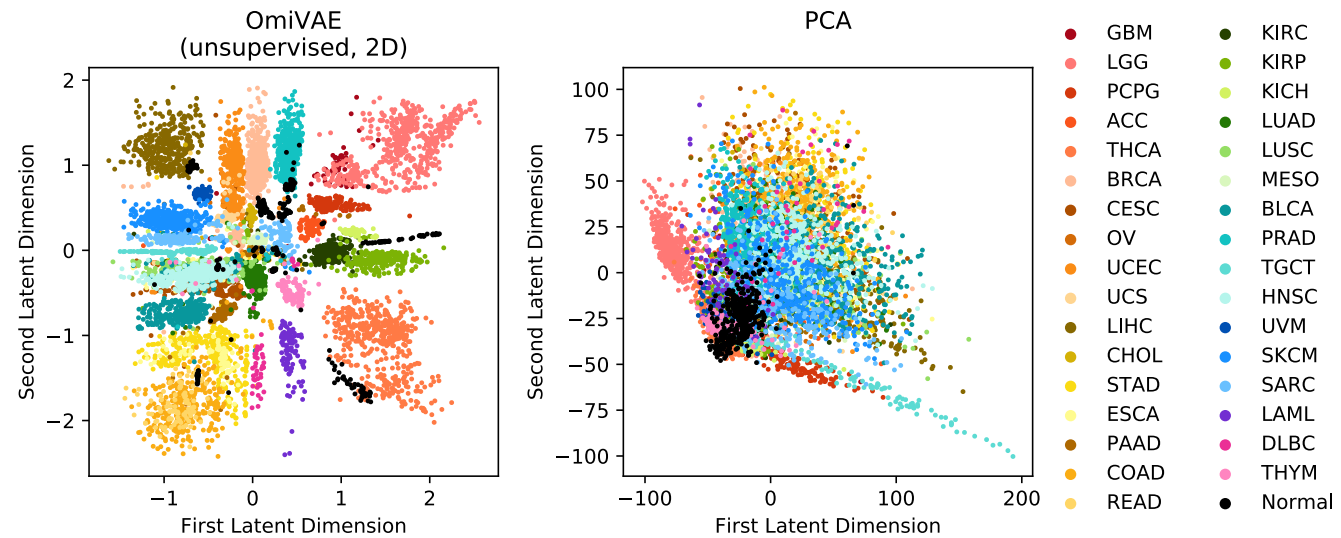
可以明显看到其要优于PCA。
再进一步拿出单独的几个癌症来看:
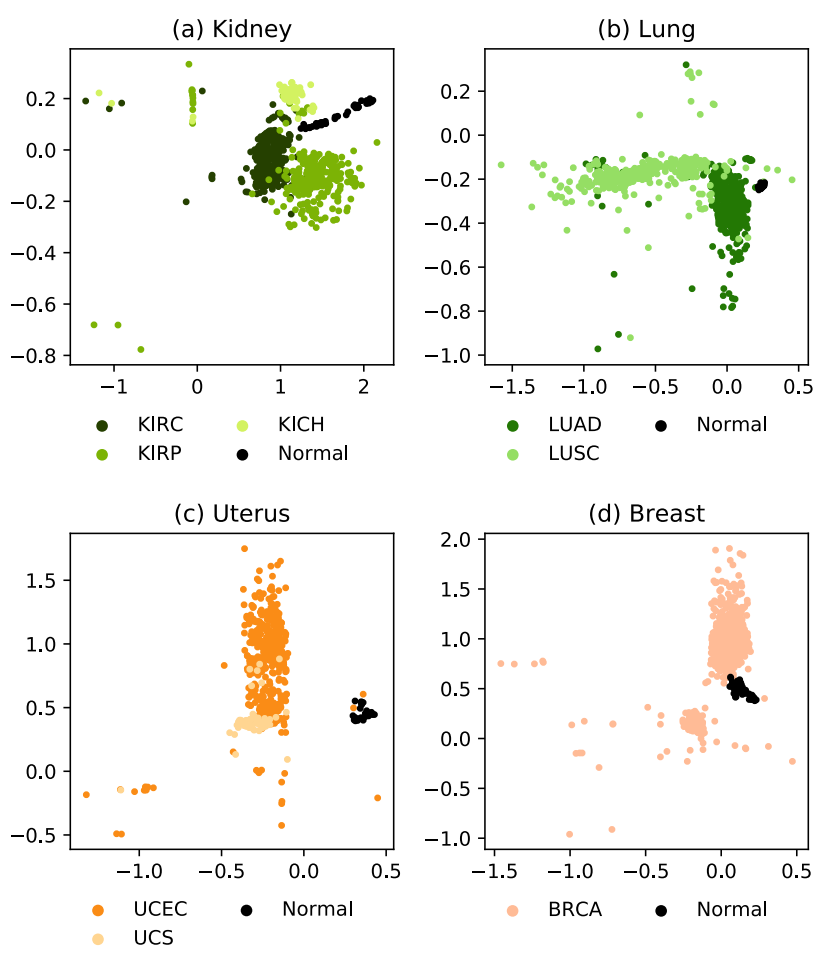
结合上面的图像,可以看到以下结论:
- OmiVAE不止能够将各个癌症类型分开,而且对于癌症的亚型,也能分开。
- OmiVAE也能够将癌症和对应的normal sample分开。
- 从整体上来看,对应癌症的normal samples是和对应的癌症样本在一起的,然后在局部他们再分开。
为了评价VAE提取的特征是否优于其他方法,这里进行实验:
- 比较的方法有PCA、kernelPCA、t-SNE、UMAP。
- 都降到2维,然后使用rbf-SVM进行34类分类任务,结果如下表所示:
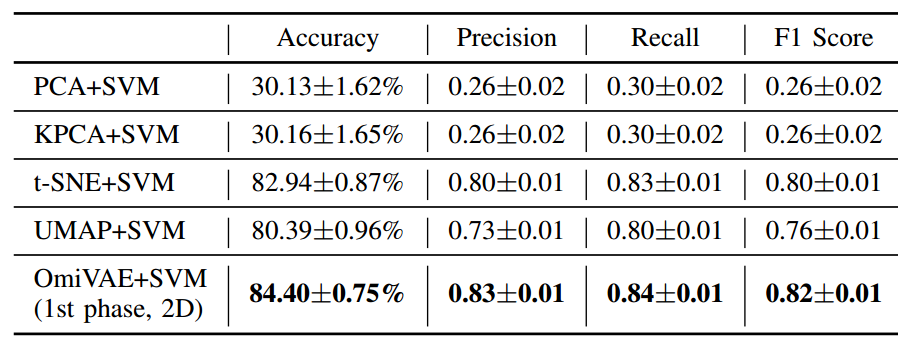
可以明显看到OmiVAE的优势。
这里最接近OmiVAE的是t-SNE,但其还有以下另外两个缺点:
- t-SNE非参数的,无法应用到其他数据集;
- t-SNE只能降维到2-3维,对于更高维度的降维不适用。
监督过程
为了保留更多的信息,这里将瓶颈层维度设为128进行。
这里比较了一下end-to-end的OmiVAE和使用瓶颈层特征+SVM的效果差异。首先是34类分类任务:
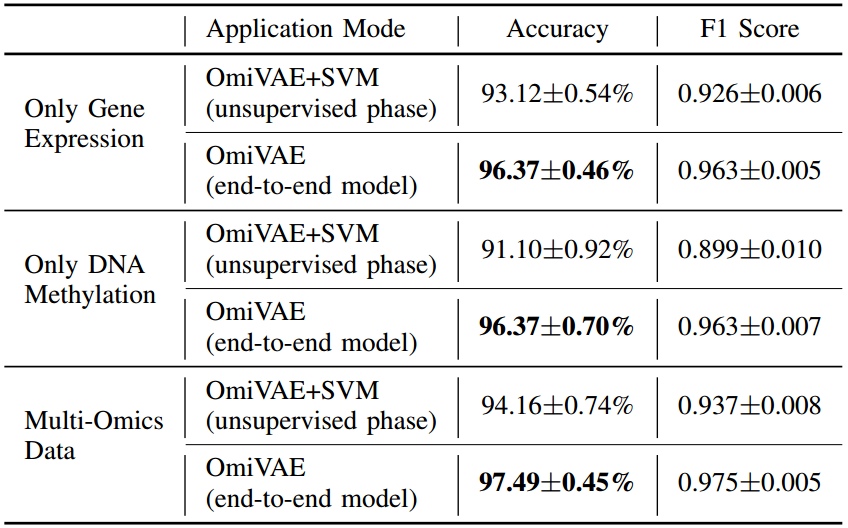
以上结果超越了之前同类研究的表现【18】。
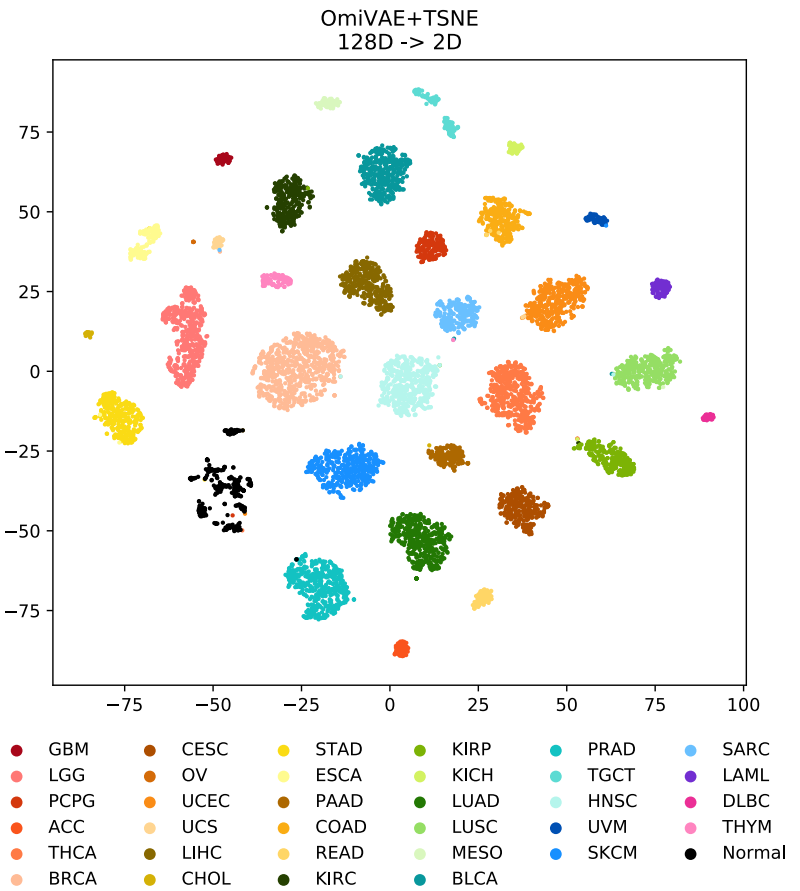
另外,128维特征(经过了监督过程微调)使用t-SNE进一步可视化,发现各个癌症类型可以比较好的分开,说明了微调过程使得特征学习偏向了癌症类型分类:
同样的,也进行了33类分类任务的比较,效果如下图所示:
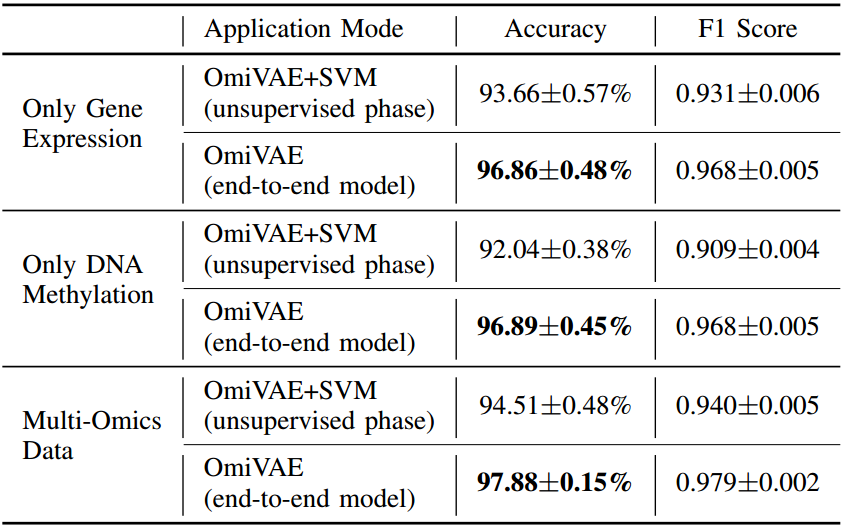
其结果也超越了之前的研究结果(【16-18】)。
Conclusion
没什么营养。
Questions
文章最大的亮点,在我看来,是利用染色体分组来减少参数量,这同样增加了一定的生物学先验信息到模型中。那通过进一步考虑基因等在染色体上的相对位置,是否有更好的效果能?
- 将Y染色体的基因去除是否不妥,其存在可能会对男性高发癌症(前列腺癌、膀胱癌、肺癌等)起到积极的意义。
- gene expression去除变量的数量有点对不上?
- 文章中只提到了beta值的设置,但alpha值没有提?



