此部分的内容来自UCSC Xena官方提供的视频教程,大家也可以到YouTube自行观看。此教程共分为3个部分:
- 第一部分是一个对Xena的简单的介绍(时长大约5min);
- 第二部分是利用TCGA中的LGG和GBM数据来做一个简单的操作演示(时长大约17min),经过第二部分的学习就能够掌握Xena可视化的大部分操作;
- 第三部分是一个时长55分钟的报告(第二部分是此部分的一个节选片段),详细介绍了UCSC Xena解决的问题、其所包含的公共数据库有哪些、利用公共数据库进行可视化分析、利用下载的数据(或私人数据)进行分析等等。
本篇内容主要来自于第二部分和第三部分。主要是如何基于公共数据库使用UCSC Xena进行简单的可视化分析。其中关于数据库的介绍,会另外单独辟出篇章进行。公开数据库的示例数据集是TCGA-LGG和TCGA-GBM的chr1和chr19的copy number,还有TP53、ATRX的mutation。私人数据的演示将放在下一篇 中进行。
数据的载入
1. 进入UCSC Xena Functional Genomics Explorer:
以下是我们主要使用的界面:
![[Functional Genomics Explorer]](/2020/05/24/tools/ucsc/ucsc-demo1/ucsc-demo_2020-05-24-15-02-58.png)
在这个界面内,数据以spreadsheet为单位展示。其可以包含单个phenotypic变量,也可以是某个gene上的DNA methylation或mutation、某条染色体上的copy number、1个或多个gene expression。其每一行表示一个样本,所要分析的所有样本叠加在一起组合成headmap(或者其他),不同种类的数据在spreadsheet上会有不同形式的展示。关于spreadsheet更加详细的介绍请见。
一般来说,我们至少需要添加3个spreadsheets(sample datasets、一个phenotypic变量、一个genomics变量)才能进行一般的genomics分析,所以页面会提供一个3-steps的wizard。
2. 选择一个合适的study(第一步)
这里实际上就是加载第一个sreadsheet--sample datasets。TCGA等公开数据库中的数据根据不同的研究目的(针对特定癌症、或者泛癌分析等)进行了打包,选择特定的研究就会载入其相关的所有样本:
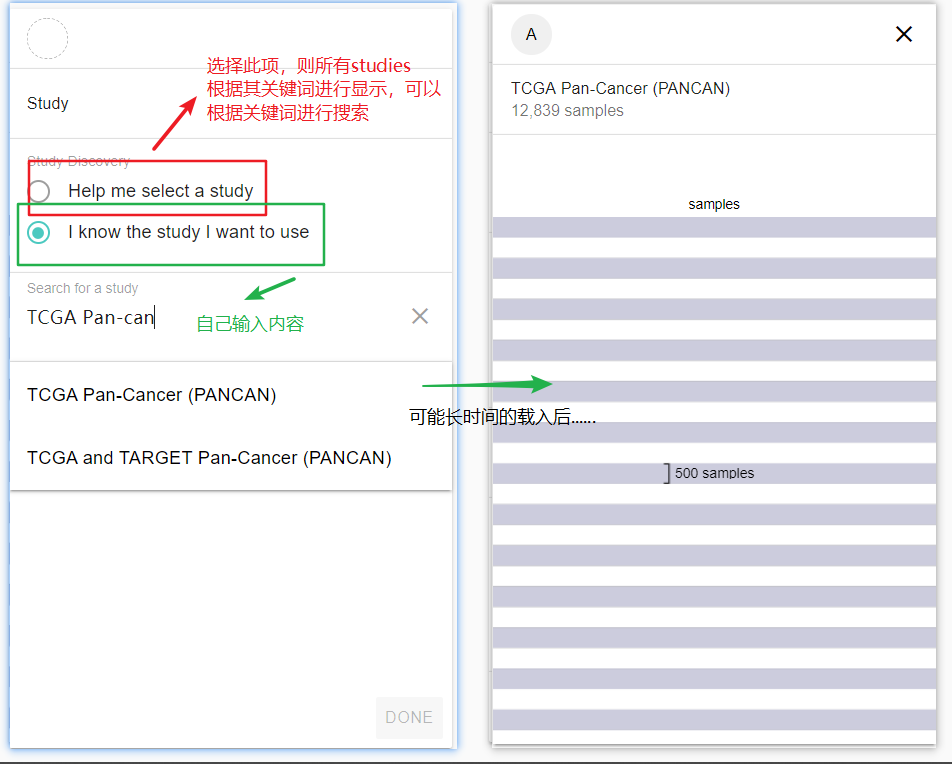
使用条带来表示样本量大小,并标注一个条带代表了多少个样本(上图中是500)。
3. 选择研究的第一个“变量”(第二步)
第二步需要我们添加第二个spreadsheet,一般来说这都是一个临床信息变量,即phenotype。这里根据我们的示例任务,我们选择“癌症类型”。这属于phenotypic类型(即临床信息),如下图所示:
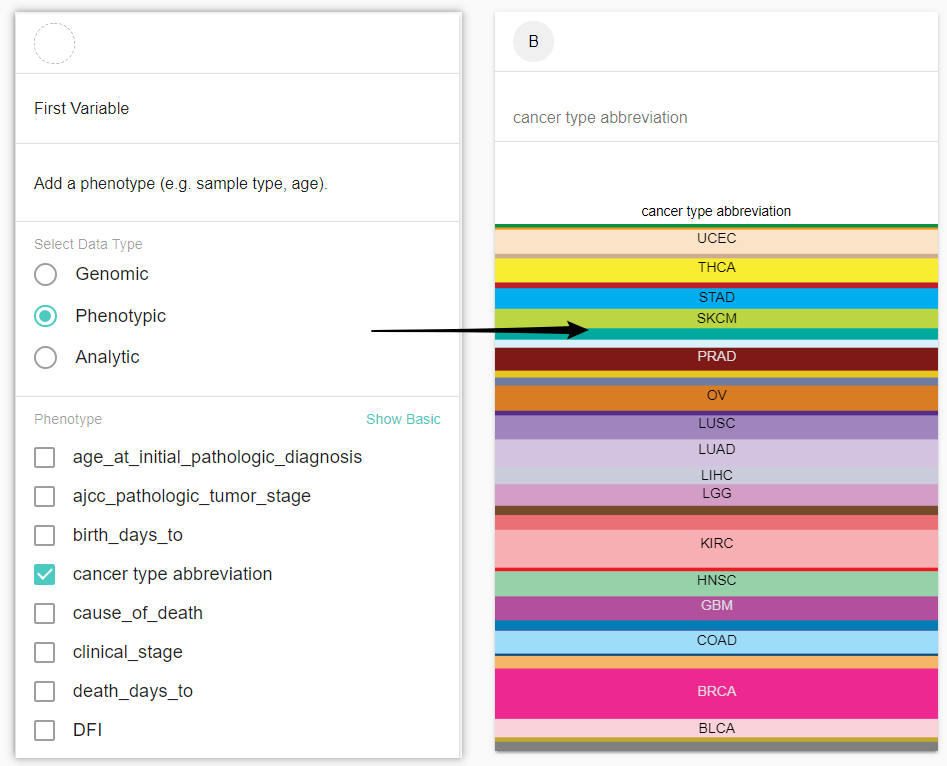
当然,我们没有必要只增加一个phenotype,如果我们的分析需要更多的phenotypes,则添加继续添加即可。当增加到3个spreadsheets的时候,界面便会跳转到分析界面上,但这没有关系,在此界面上可以继续添加spreadsheets。我们也可以在一次添加中添加多个变量,只需要同时勾选多个phenotypes即可。
变量类型
变量类型一共有3种,除了上面提到的phenotypic,还有另外两种类型,genomics和analytic,genomics将在下一步进行介绍,这里简单介绍一下analytic。
analytic型变量,就是基于genomics数据,通过一些数据分析方法得到的诸如分子亚型、评分等数据,比如免疫亚型:
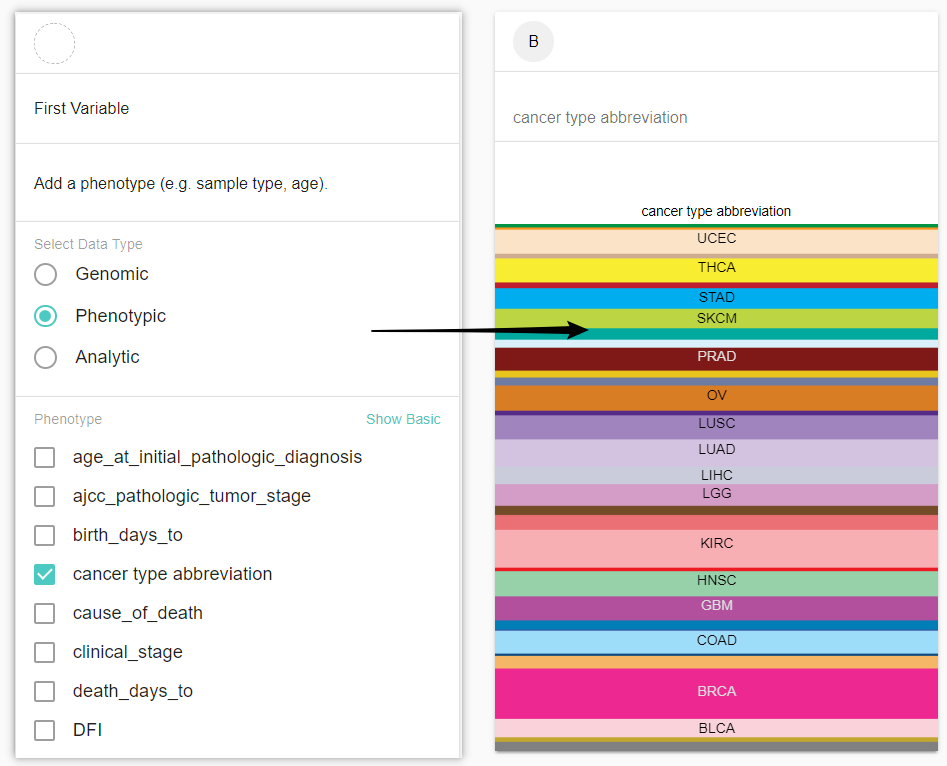
3. 选择第二个“变量”(第三步)
一般是genomics类型。
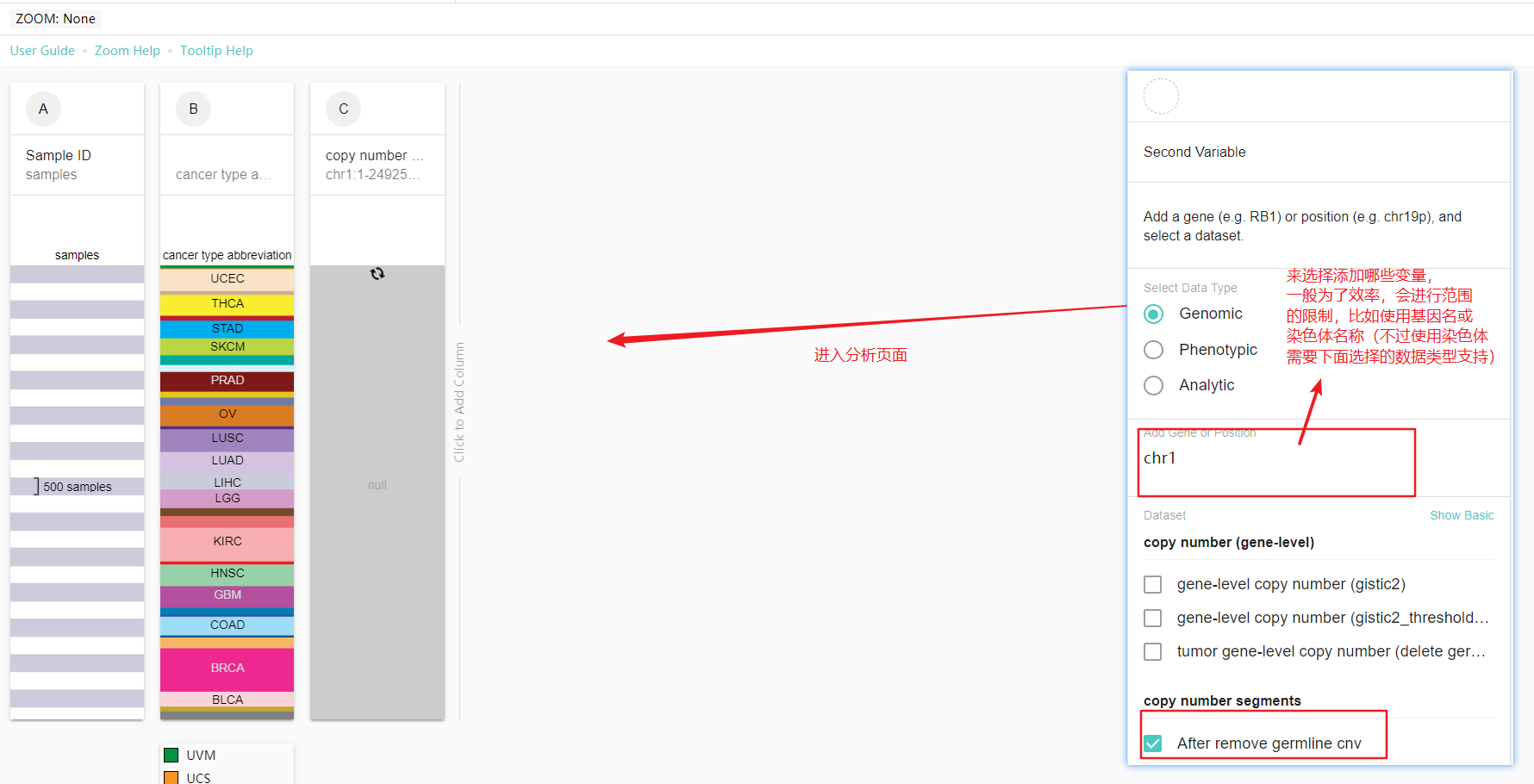
数据分析
整个分析界面如下面所示。实际上挺简单的,图形界面操作,看一眼也都能知道是怎么用。
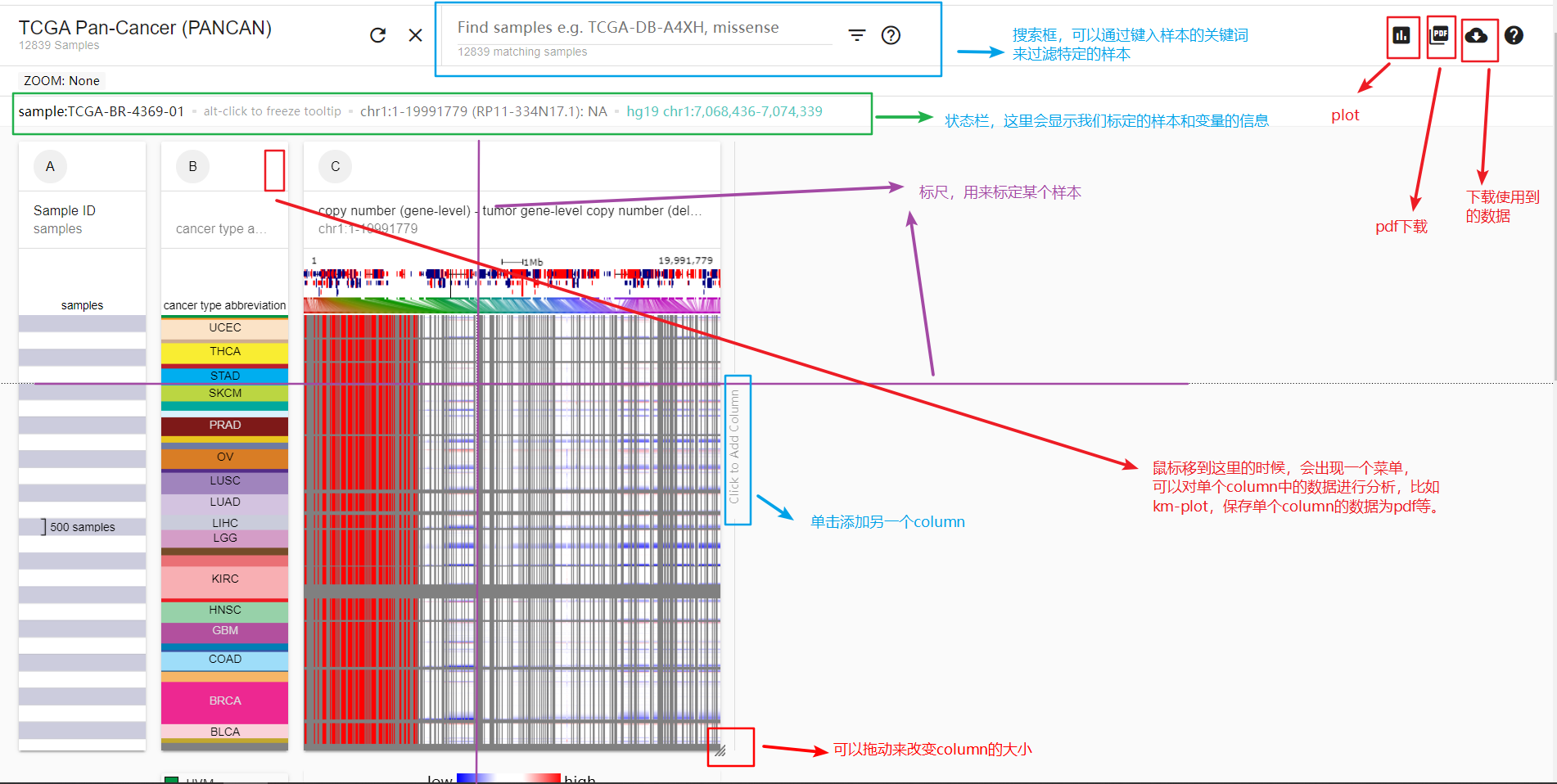
1. 继续添加剩下的chr19数据和mutation数据

我们可以看到,连续性的变量主要以heatmap的格式存在,而至于分类变量则是以不同颜色的条带来表示。很多genomics的数据,拥有更多的信息(比如copy number、methylation等),则会给出更多的信息,比如每个位点所在基因或染色体的位置、状态等等。总之,信息量是很丰富的。
2. 筛选出我们需要的数据
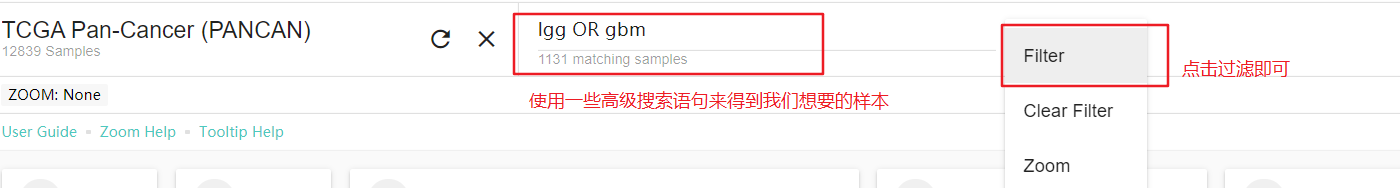
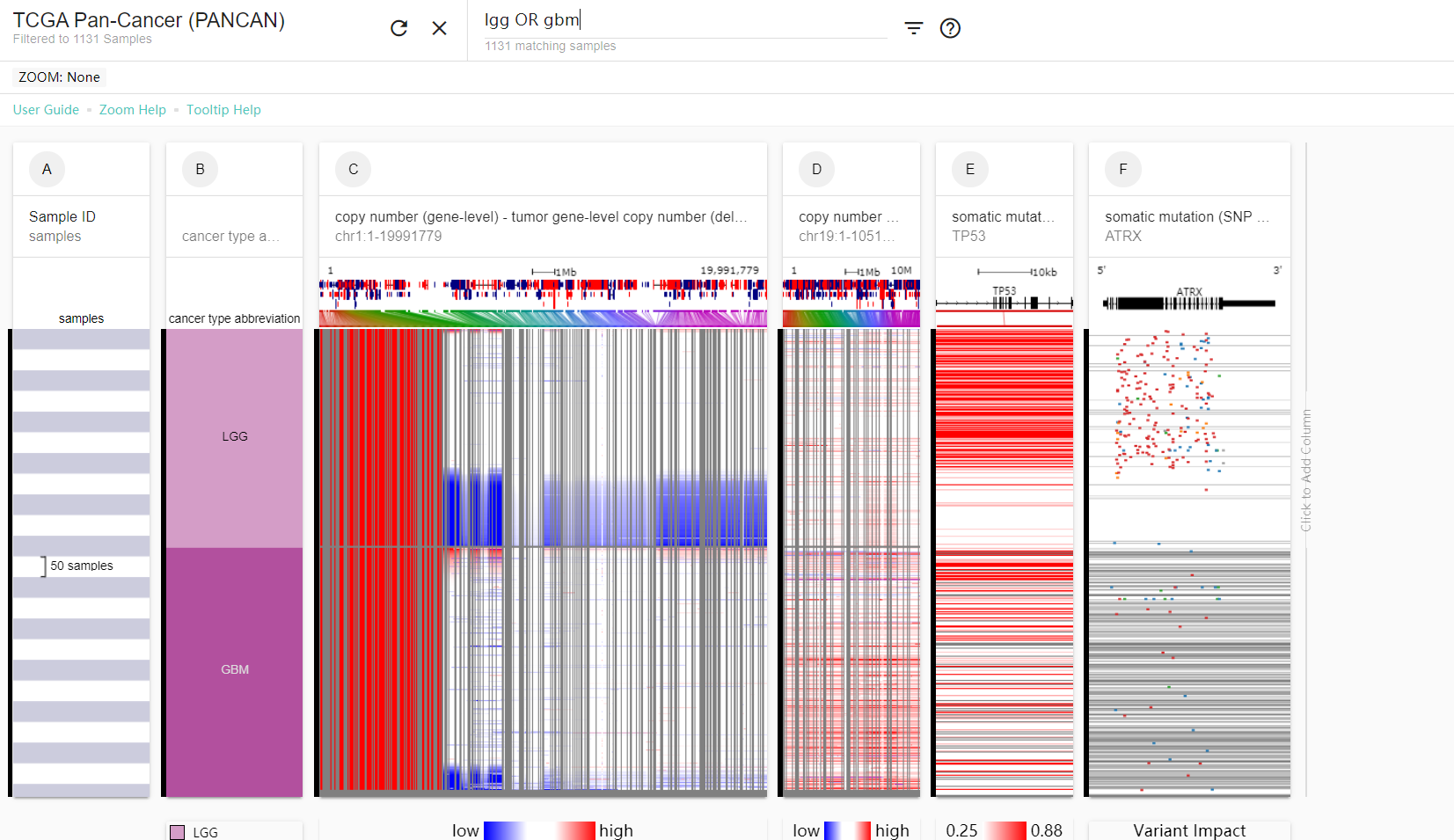
得到下面的结果:
实际上我们可以通过移动spreadsheet来实现样本按照不同的类型进行排序。比如我们可以将TP53的mutation移到前面去,则样本会优先按照tp53的值进行排序,然后才是cancer type:
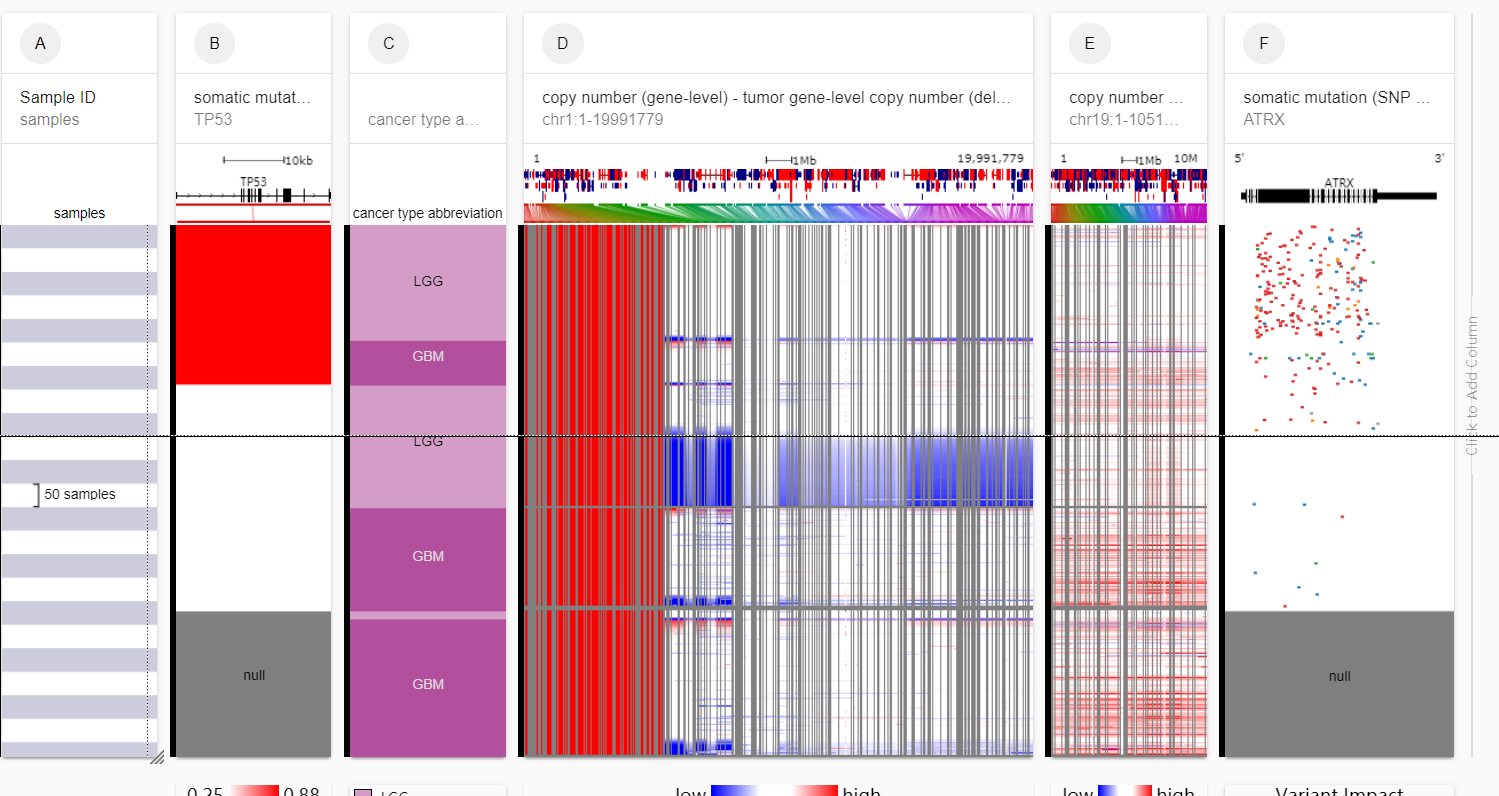
我们可以看到有大量的null(缺失样本),我们不希望要这些样本,则可以继续使用上的样本搜索框来实现过滤(这里把tp53的expression数据类型改成了mutation,之前只是为了方便说明不同类型的变量的展示结果):
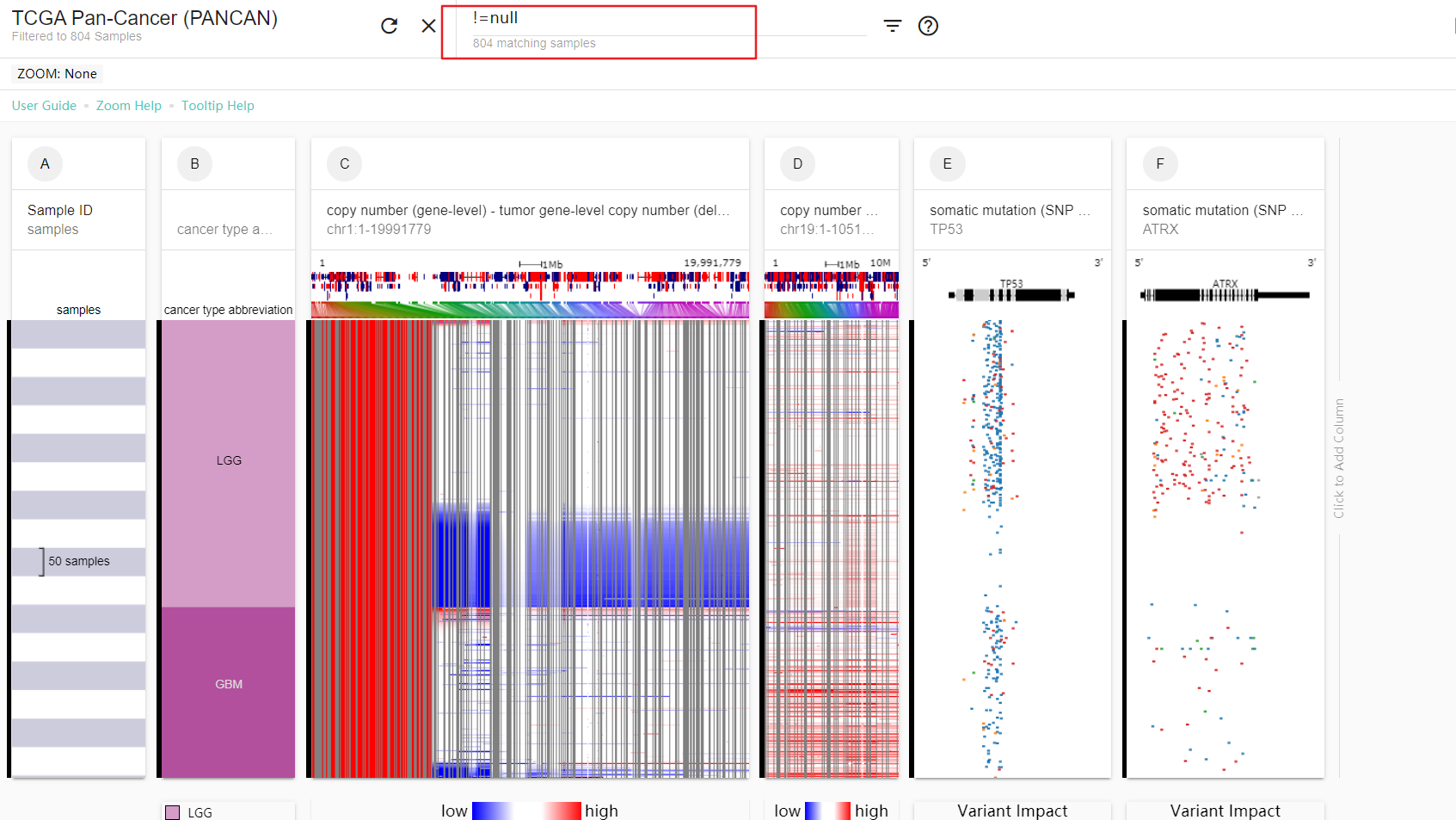
3. 简单的数据分析
这一部分的图可能和上面的不太一样,进行上面的内容的时候,网不是太稳定,所以只能加载一段染色体上的copy number数据,而本部分是加载了1号和19号染色体的全部copy number数据,所以才能够比较直观的看出差异来。
实际上根据这些可视化内容,我们就已经可以进行一些简单的分析了:
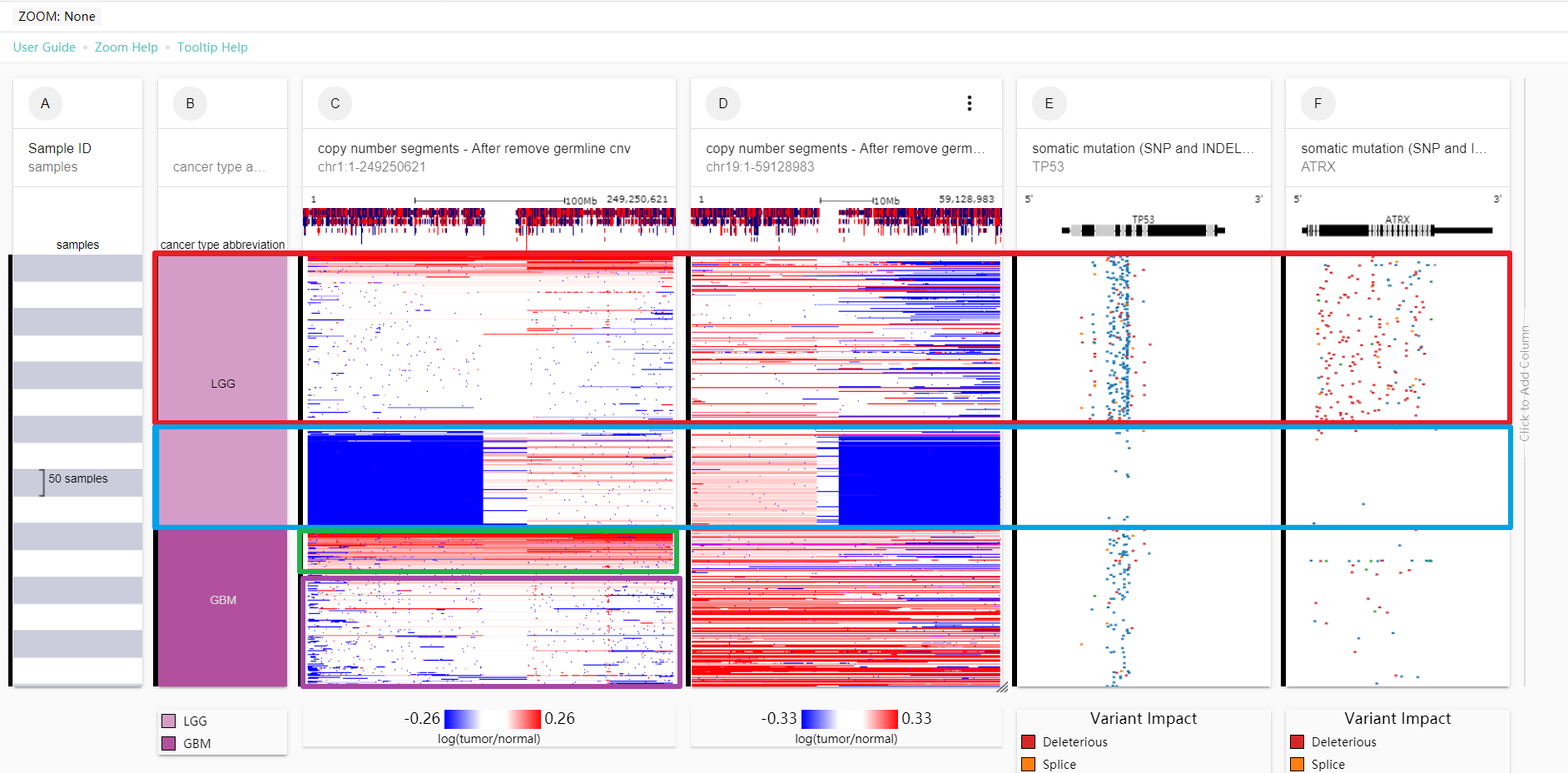
可以看到,根据1号和19号染色体的copy number,可以很明显地将LGG的样本分为2个部分,这两个部分在TP53和ATRX mutation上也存在着明显的不同。
而在GBM上,则无法看到这样明显的区别。不过基于1号染色体的copy number数据,可以看到GBM样本也可以进一步分为2个小的亚型。
4. 绘制两种癌症的K-M plot
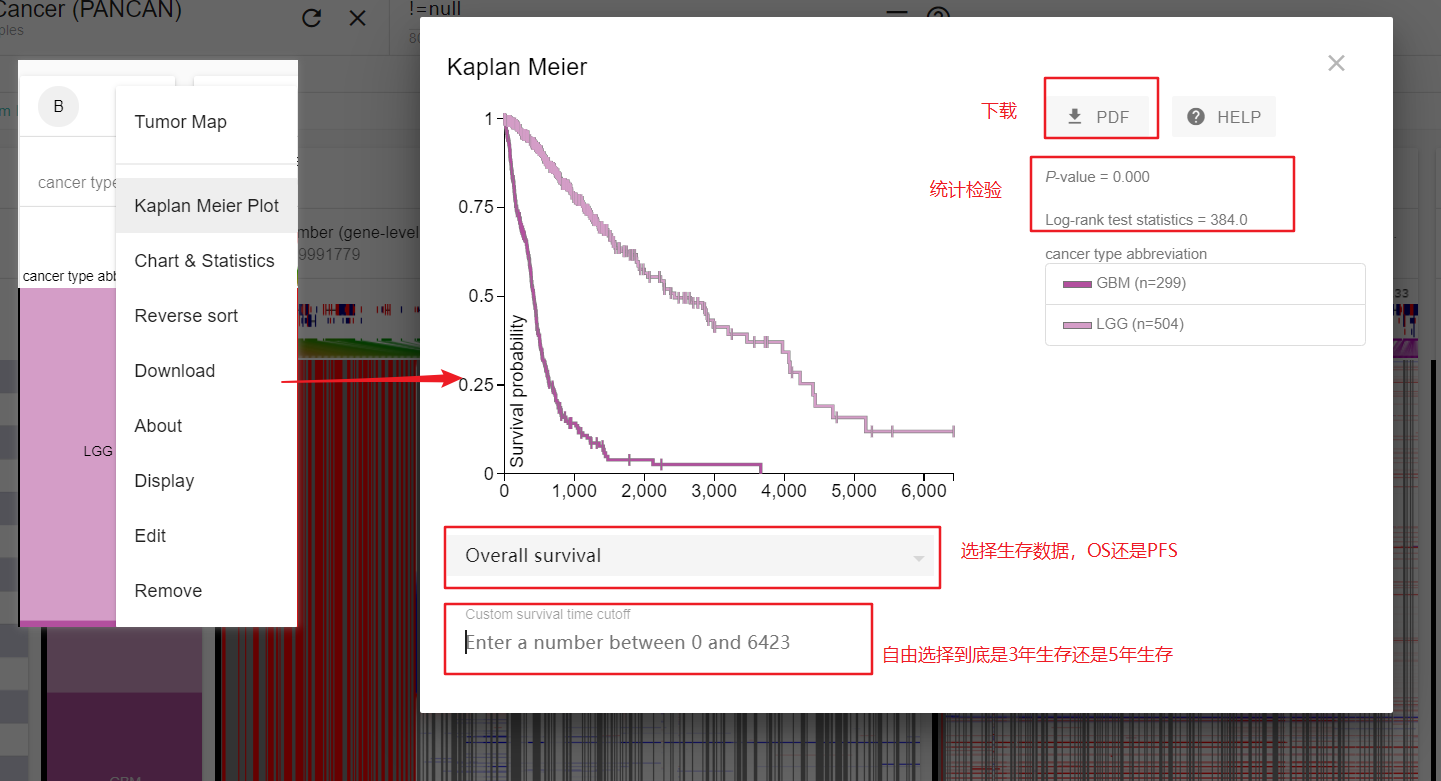
详细的关于K-M plot的信息,请见。
5. 对数据进行statistic可视化
点击右上角进入chart页面,可以绘制一些统计图示,从整体上来对数据进行一定的分析。(不是所有数据都可以绘制chart,比如mutation数据就不行)
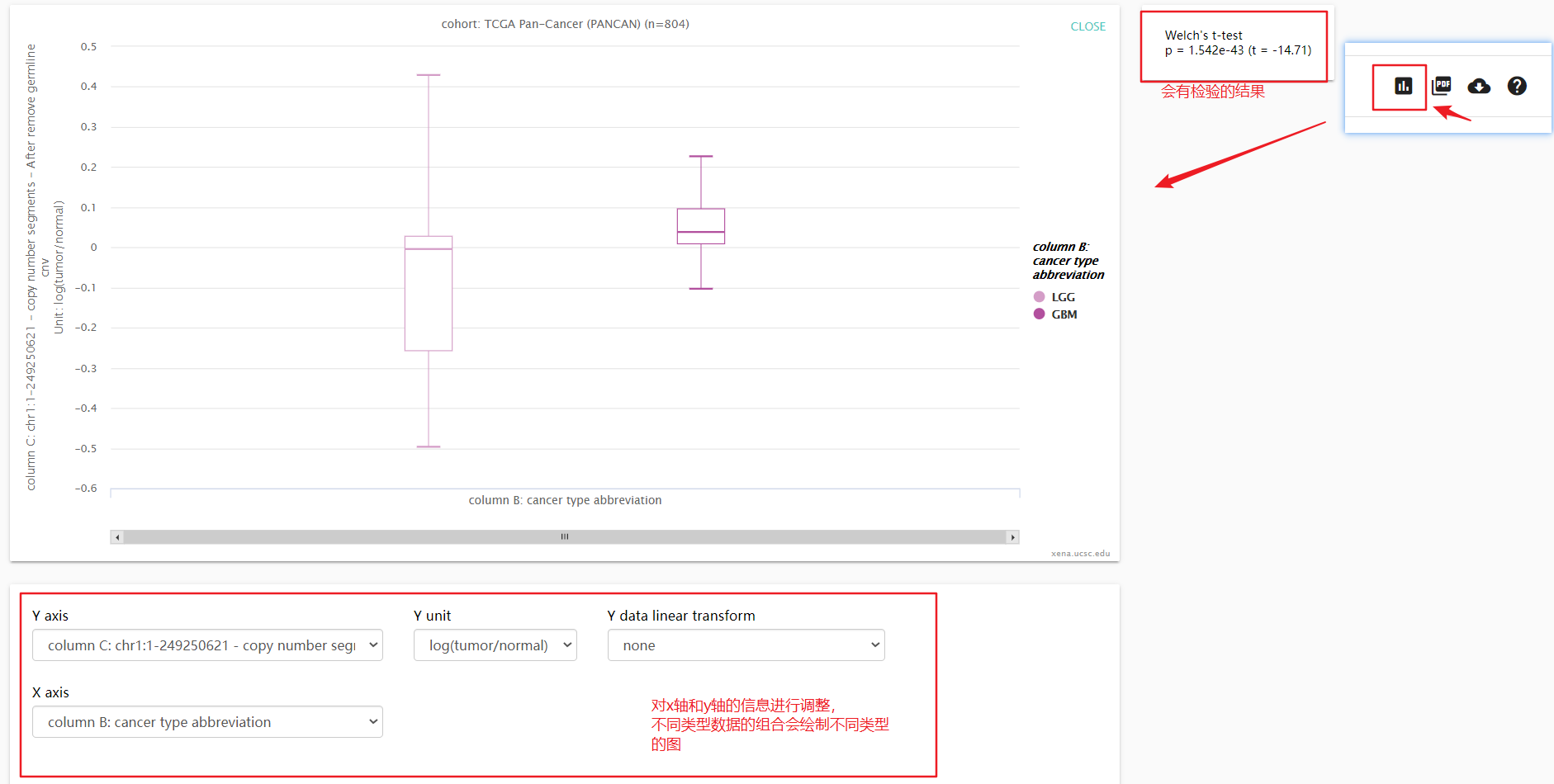
关于chart的详细信息,请见
暂存当前的分析结果(Bookmarks)
有两种方式:bookmarks url和export files(但都只能保存30天)。
- Bookmarks url,将当前的内容保存到云端,会生成一个url,用来进行分享。为了安全起见,私人数据在保存的时候会被剔除。
- export会将当前可视化所需要的所有内容都整理成一个json文件,保存到本地,当使用时,再通过import导入即可,这种方式可以将私人数据的可视化保存。
其他
以上图示结果都可以下载为PDF文件,比较方便。
这基本上就是UCSC Xena的所有功能,功能不算太多,但重要的在于可以马上形成漂亮的可视化。在相关研究的计划阶段,可以使用这些功能快速的验证猜想,有助于之后的研究设计和决策。



