经过,我们能够对TCGA等公开数据库进行简单的可视化处理了,但实际上UCSC Xena可以做到更多。
这里我们从GEO下载数据集GSE121720作为私人数据集,然后使用UCSC Xena进行分析。这一部分的tutorial可以见官方提供的视频教程,或者是其网页。
1. 安装Local Xena Hub
为了安全性和服务器压力考虑,对于私人数据的处理,其服务器是使用Local Xena Hub软件架设在本地机上的。Local Xena Hub由java编写。
下载需要点击VIEW MY DATA,这里提供windows的直接下载链接。在windows上的安装比较简单,就是点一点就好了。
2. 开启Local Xena Hub
直接进入VIEW MY DATA,如果已经安装了Local Xena Hub,并且其安装位置在Applications folder中(其实自动就安装到这里),则浏览器会自动开启它。如果没有自动开启,需要主动双击打开exe文件。这时候网页会连接到本地服务器。
需要使用Chrome或Firefox浏览器。
3. 清理数据集
为了本地服务器可以识别私人数据集,我们必须把数据集整理成一定的格式。
总的来说有以下几条要求:
- 首先,本地服务器可以识别多种数据,但这些数据必须是TCGA的“level 3”格式的数据(比如gene表达的值、transcript的值、probe的值)。不支持FASTQ、BAMs等更加“raw”的数据格式。
- 所有的数据必须是tab-delimited的,文件的后缀一般是
.tsv或.txt,不支持Excel格式(.xls或.xlsx)。 - 不要有重复的genes/probes/identifiers或samples,不然本地服务器只会保留重复的第一个。
- 使用空格或
"NA"来表示缺失值。 - 使用
.来表示小数点,不要使用,(在一些语言中用逗号做小数分割符)。
至于具体的数据格式,这里细化分为了4种,分别进行介绍:
3.1 Basic Genomic data
大部分的基因组level3级数据,是正常的numerical rectangle/matrix/spreadsheet型数据。即每一列是一个样本,每一行是一个变量,反过来也可以。比如下面的这些:
- RNA-seq expression (exon, transcript, gene, etc)
- Array-based expression (probe, gene, etc)
- Gene-level mutation
- Gene-level copy number
- DNA methylation
- RPPA
- and more ...
图示(注意Sample的存在):
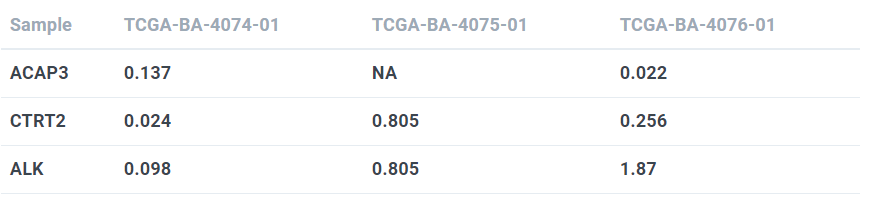
服务器可以自动将一些probesname(比如TP53、EGFR等)。其支持的probes中可以查到。
3.2 Basic Phenotypic data
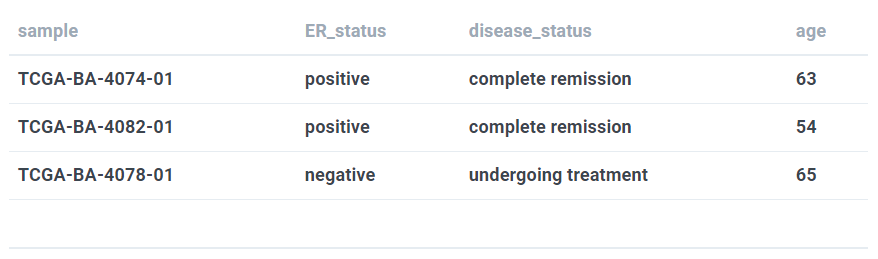
也是rectangle/matrix/spreadsheet型数据,用来描述临床信息,可以是categorical或numerical的。可以是一行是样本一列是变量或者反过来,比如下面的这些:
- phenotype/patient/clinical data (age, weight, if there was blood drawn, etc)
- sample/aliquot data (where it was sequenced, tumor weight, etc)
- derived data (regulon activity for a gene, etc)
- genomic signatures (EMT signature score, stemness score, etc)
- other (whether a sample has an ERG-TMPRSS2 fusion, whether a sample has WGS data available, etc)
这一类数据是最灵活的,比如下面的形式:
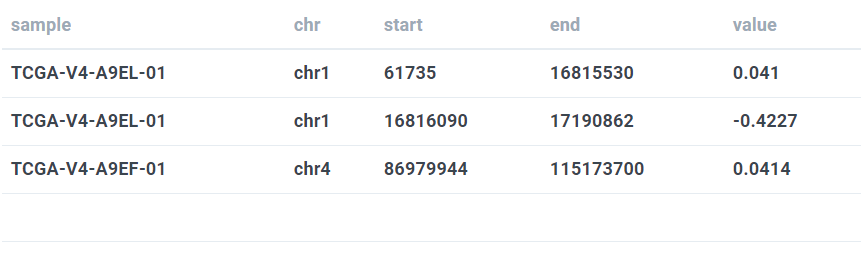
3.3 Advanced Segmented data
需要有5列,分别是sample、chr、start、end、value,现在支持的coordinators是hg38、hg19和hg18。主要就是copy number。
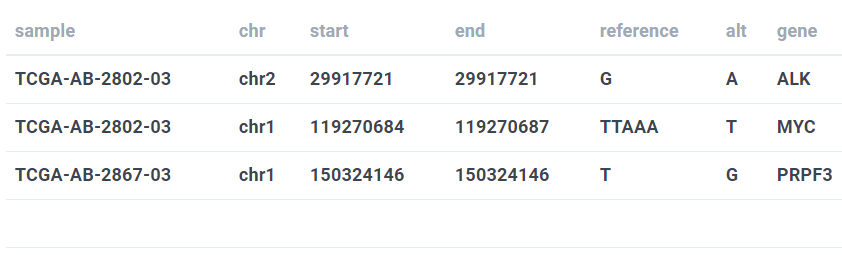
3.4 Advanced Positional data
需要有6列,分别是sample、chr、start、end、reference(参考基因组)、alt(变异后的编码)。另外还可以有的列有gene、effect、DNA_VAF、RNA_VAF、Amino_Acid_Change。这些列不是必须的但会增强数据的可视化(比如根据effect为不同的突变赋予不同的颜色)。
现在支持的coordinators是hg38、hg19和hg18。
3.5 其他
也支持其他数据,但需要和官方联系得到支持。
现在我们下载的数据是这样的格式:
也需要进一步的处理才会符合上面的要求,以下是处理代码:
# py preprocess.py
import os.path as osp
import re
import pandas as pd
data_root = "G:\\dataset\\UCSCexamples"
cli_fn = "GSE121720_series_matrix.txt"
gene_fn = "tpm.txt"
# 处理clinical 数据
cli_data = {}
with open(osp.join(data_root, cli_fn), "r") as f:
for line in f:
if line.startswith("!Sample"):
# 1. 将每一行的第一个元素去除!Sample_作为key,剩下的提取出“”中的部分作为value
key, value = line.split("\t", maxsplit=1)
key = key[len("!Sample_"):]
value = re.findall(r'"(.*?)"', value)
# 2. 如果value中的元素以xx:yy形式存在,则xx作为其新的变量名
if all([":" in v for v in value]):
v1s, v2s = [], []
for v in value:
v1, v2 = v.split(":", maxsplit=1)
v1s.append(v1.strip())
v2s.append(v2.strip())
if len(set(v1s)) == 1:
key = v1
value = v2s
# 保存到dict中
cli_data[key] = value
cli_data = pd.DataFrame(cli_data)
# 这里变量名是title,需要对其进行一些处理:包括去掉一些冗余的字符和改一下变量名
cli_data.insert(
0, "Sample", cli_data["title"].str.split(" ", n=1, expand=True)[0])
cli_data.pop("title")
# 把名称超过255长度的列去掉
bool_index = cli_data.columns.map(lambda x: len(x) < 255)
cli_data = cli_data.loc[:, bool_index]
cli_data.to_csv(osp.join(data_root, "clean_cli.tsv"), sep="\t", index=False)
# 处理RNAseq data(主要为第一列加Sample的column name)
writer = open(osp.join(data_root, "clean_rnaseq.tsv"), "w")
with open(osp.join(data_root, gene_fn), "r") as f:
for i, line in enumerate(f):
if i == 0:
line = "Sample\t" + line
writer.write(line)处理结果:
4. 载入数据集
载入数据集的方式有两种:
4.1 使用VIEW MY DATA网页载入
直接使用网页上的3-steps wizard来完成,这需要一个文件一个文件的import,对于文件比较少的比较适合。
注意事项: * 变量名长度不能超过255。 * 将表示样本的列放在第一列或第一行。
结果:
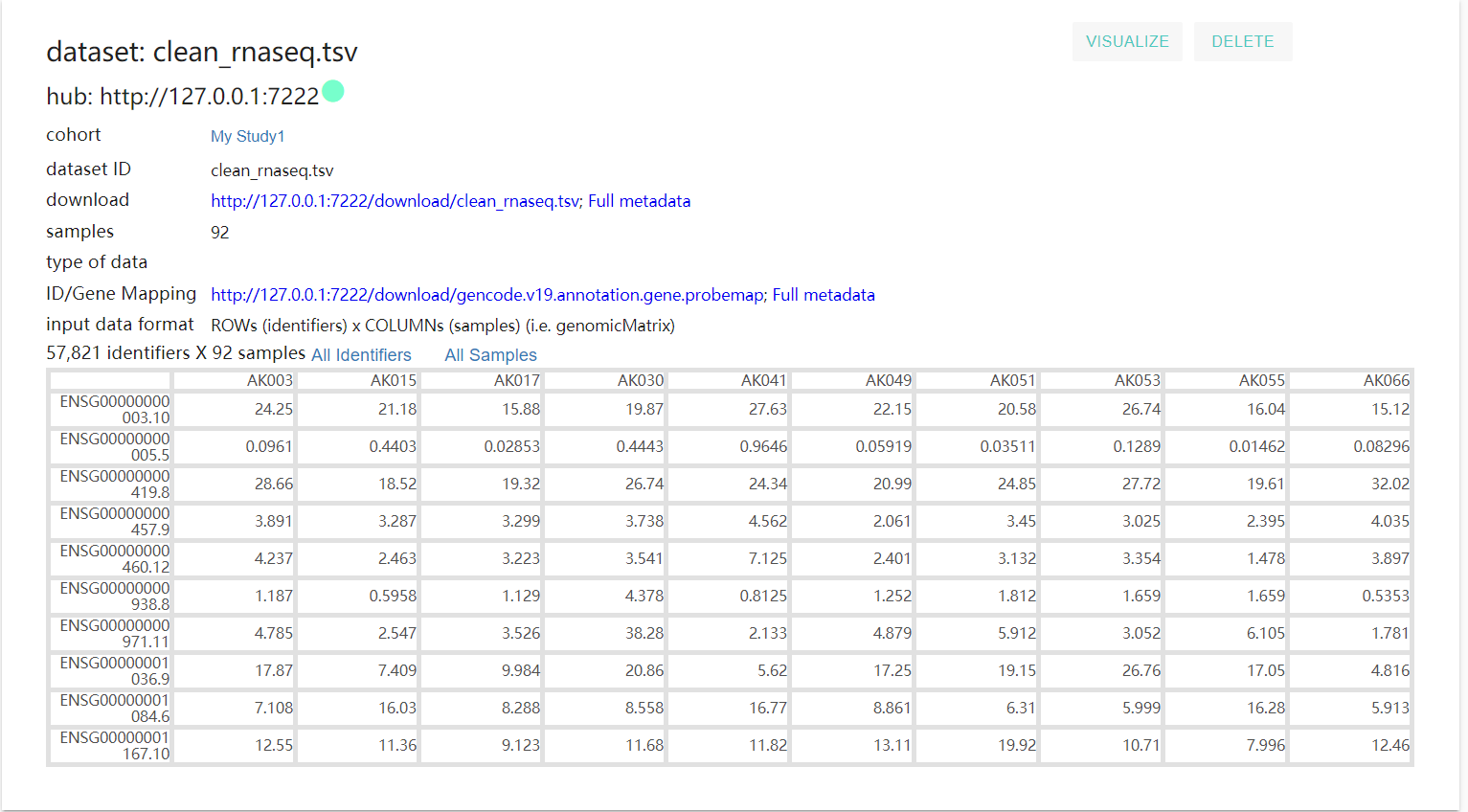
4.2 使用命令行载入
Toturial地址,这适合数据文件比较多的时候,可以通过写一个metadata file来载入。
但实际上也不是很方便,每一个数据文件就必须创建一个json文件。
Metadata file
- 需要和数据文件在同一个路径下
- 需要json文件名和数据文件名一致:
my_data.tsv--my_data.json
metadata file有以下field可以填写:
type:必填,是数据这个数据文件的类型,genomicMatrix:Basic Genomic data,必须是一列是一个样本,一行是一个变量,不支持另一种形式。clinicalMatrix:Basic Phenotypic data,必须是一列是一个样本,一行是一个变量,不支持另一种形式。mutationVector:Advanced Positional data。genomicSegment:Advanced Segmentation data。
cohort:必填,即study的名字。assembly:如果是mutation或segmentation copy number数据,需要填写,即支持的coordinations。Probemap:这个比较麻烦,简单来说,就是如果数据集使用的是自己的probes,则需要建立一个probes到其他identifiers的map,所以需要上传有这个映射关系的文件。所以需要准备3个文件:
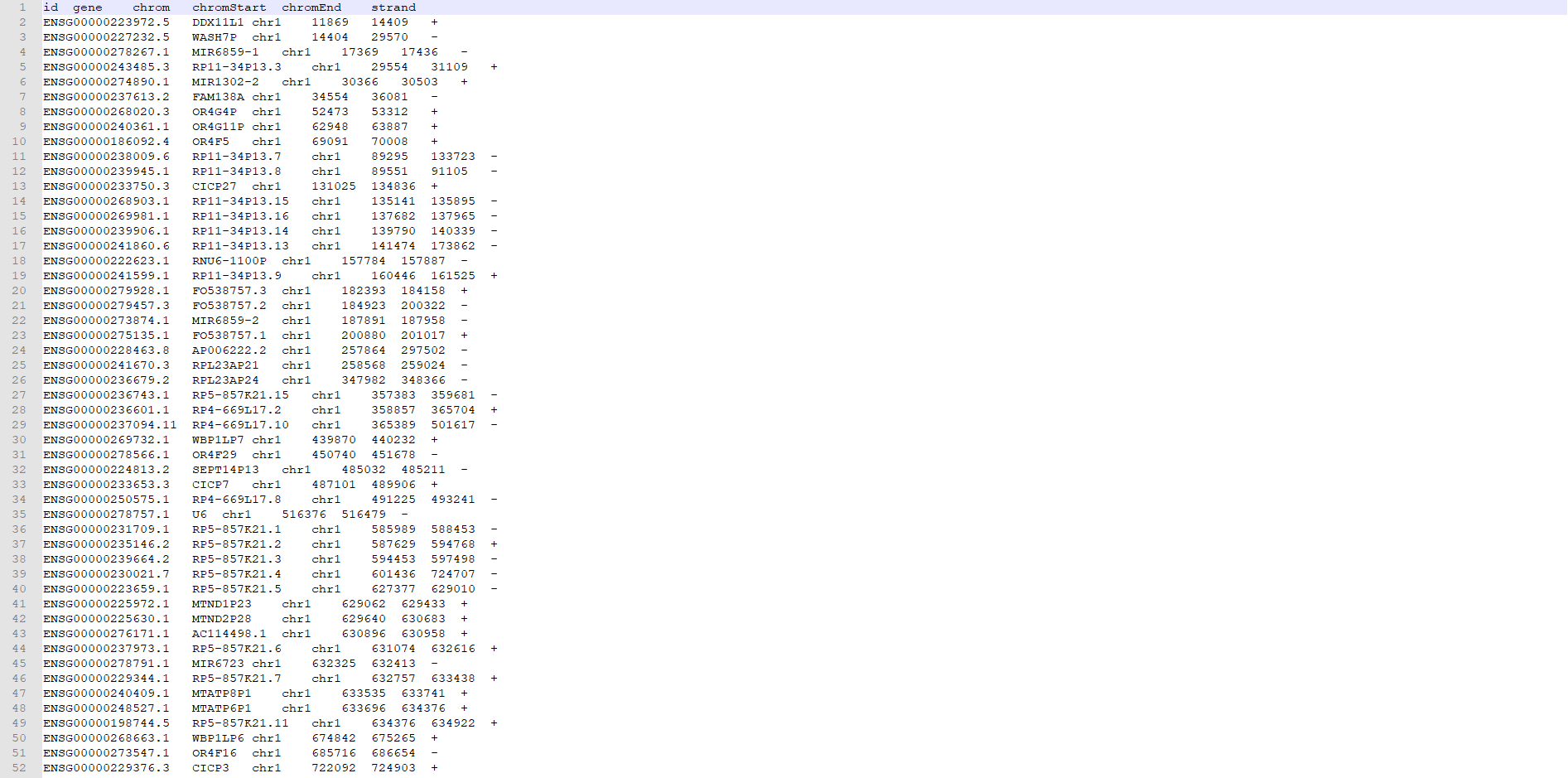
Probemap文件:比如:
probemap.json:{ “type”:“probeMap”, “assembly”:“hg19"}data.json加上如下一条:
":probeMap":"/unc_v2_exon_hg19_probe_TCGA"data.tsv
UCSC Xena官方准备了一些probemap文件,可以使用xenaPython app来获得,可见{% post_link ucsc-python %}的介绍。
命令行
上传数据:
# 上传文件下的所有数据 java -jar cavm-0.xx.0-standalone.jar -l ~/xena/files/* # 上传一个数据 java -jar cavm-0.xx.0-standalone.jar -l ~/xena/files/file1.tsv删除数据:
java -jar cavm-0.xx.0-standalone.jar -x ~/xena/files/file1.tsv java -jar cavm-0.xx.0-standalone.jar -x ~/xena/files/file1.tsv ~/xena/files/file2.tsv帮助:
java -jar cavm-0.xx.0-standalone.jar -h
需要找到那个
cavm*.jar文件,在那个文件所在路径下运行。
5. 数据集分析
其分析方式和一致,下面是最后的可视化结果:

有几点需要注意:
- 如果想要做K-M plot,则需要clinical data中有相应的Time to Event和Event变量。其命名必须按照tutorial-kmplot中进行。
- 如果要为自己的实验室或科室创建一个数据服务器,则需要另外的步骤,请见Hubs for institutions, collaborations, labs, and larger projects。



