依靠上方的搜索框,可以完成一系列数据筛选工作,甚至可以创建自己的分组。
我们可以在上面输入supported search terms,符合这些terms的样本将被高亮并使用black bar标记,然后就可以通过右侧的filter button下的不同的选择实现不同的功能:
Filter:将那些没有被高亮的样本移除。Zoom:方法这些被高亮的样本。New column:创建一个新的spreadsheet,其中被高亮的样本为True,不符合的样本为False,然后这个spreadsheet就可以用于接下来的分析(比如K-M plot等等)。可以选择spreadsheet下拉菜单中的display,将true和false改成其他的名称。
支持的search terms
分类features:某个分类变量的level,比如Stage变量的'IIA'或'Stage IIA'。
连续features:必须使用"ABC..."来进行,"A:>2"来选择大于2的样本,还有=、>=、<=、<、>、!=等。
对于mutation数据:
- 找到所有存在protein change的mutations的样本:V600E
- 找到所有功能影响是'frame'或'nonsense'的样本:D:frame OR nonsense
- 找到所有TP53带有突变的样本:TP53
- 找到所有TP53不带突变的样本:!=TP53
缺失值:
- 所有没有缺失值的样本:!=null
- 只有一列(第二列)有缺失值的样本:B:!=null
样本ID:TCGA-DB-A4XH。
A表示第一列、B表示第二列、...,A:YES表示第一列中值为YES的样本。
逻辑运算符(OR、AND),还可以配合括号来改变运算顺序:"Stage II"(B:Negative OR C:Negative),表示任何column中有"Stage II"并且第二列是Negative或第三列是Negative的样本。
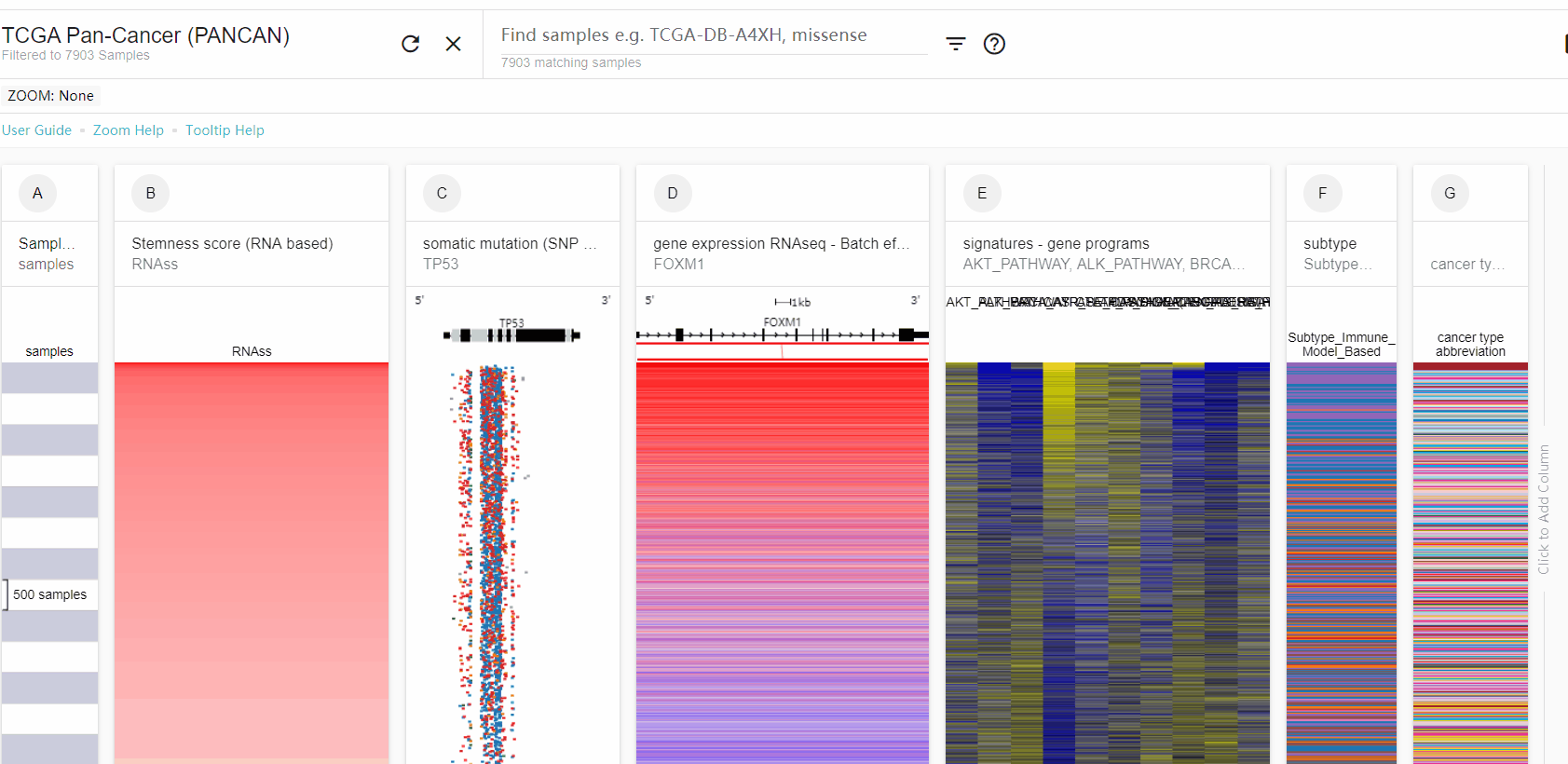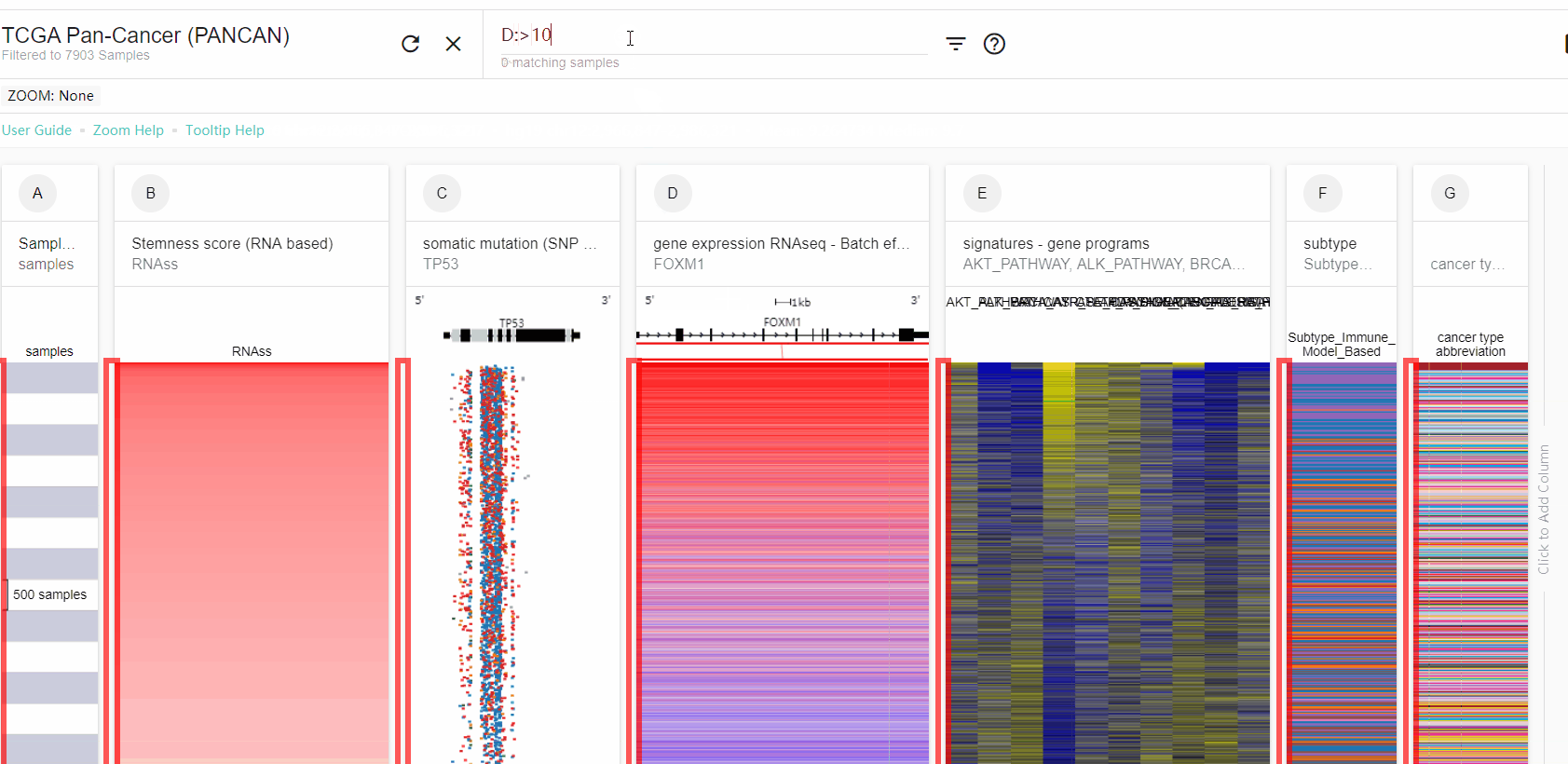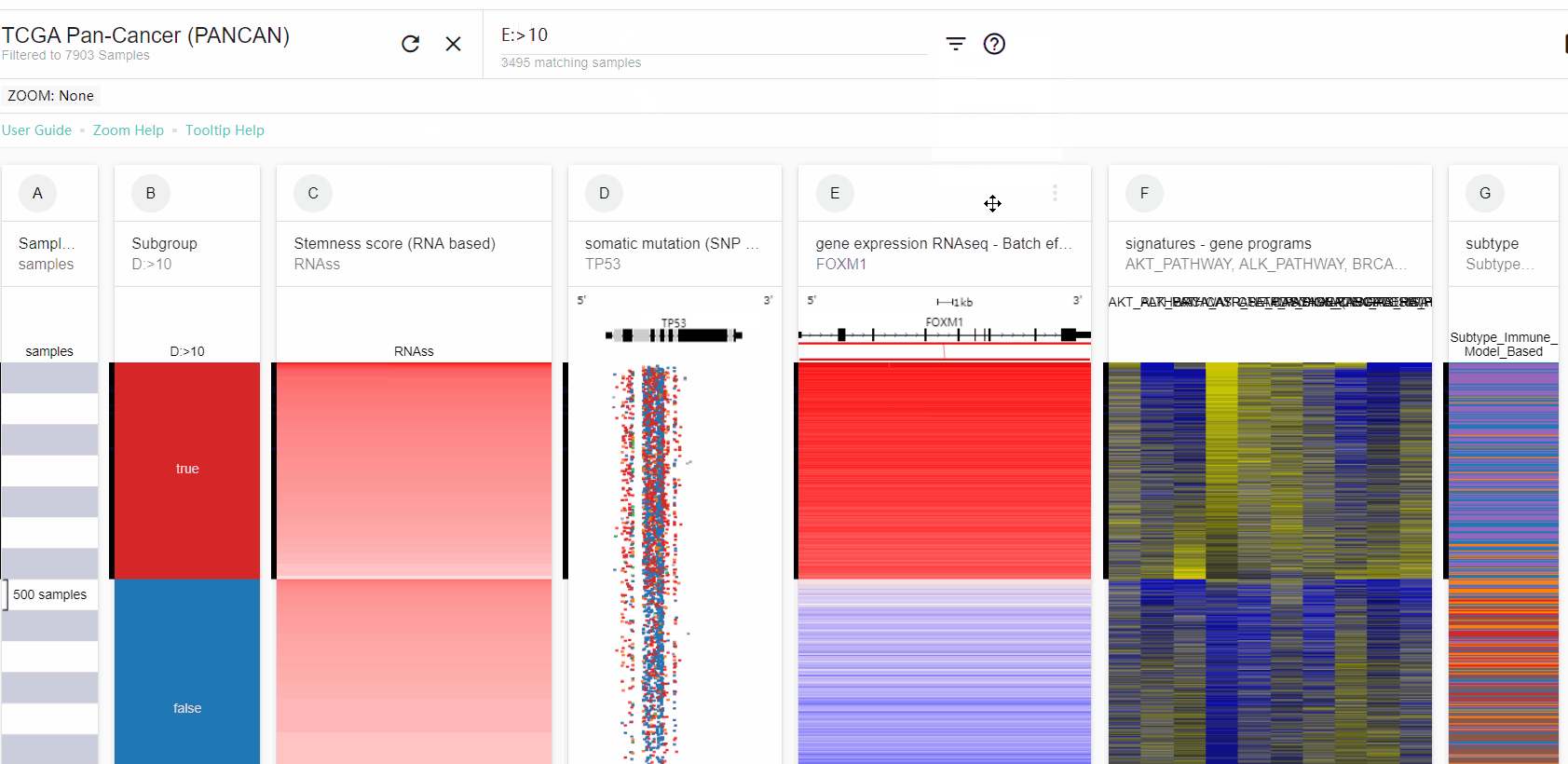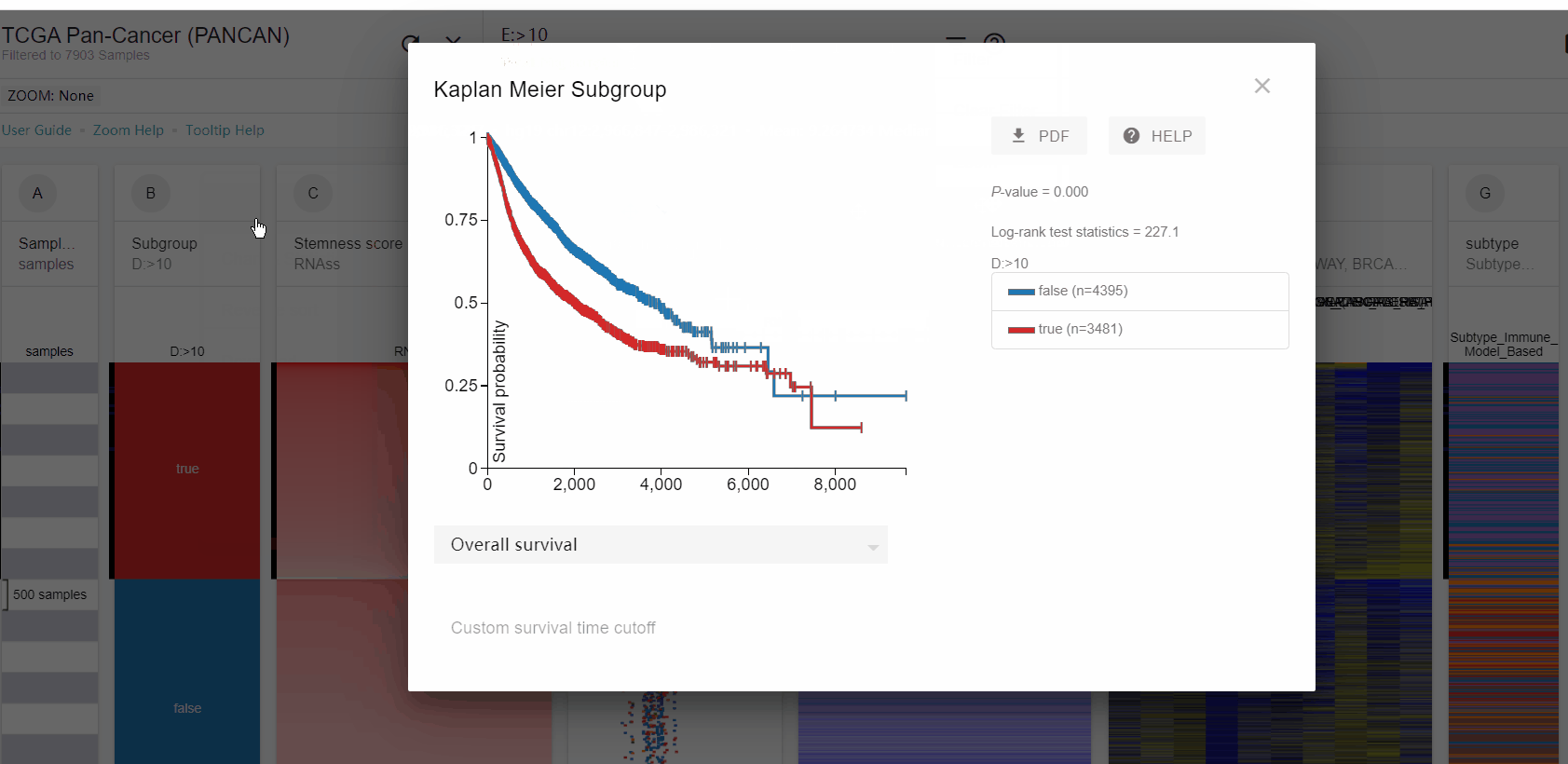
FOXM1 RNAseq表达以10为分界进行分组,并绘制K-M plot: