Classification of Cancer Types Using Graph Convolutional Neural Networks
- 杂志: frontiers in physics
- IF: None
- 分区: None
- github
Introduction
癌症早期筛检和分类有助于提高患者的生存概率。
现在很多机器学习方法被应用到公开数据库进行分析:
使用TCGA数据,K近邻进行基因选择和癌症分类,并达到了90%以上的准确率【10】
使用fully connected deep neural networks,在tumor samples(6703)和normal samples(6402)上进行训练,并评估了各个基因对最终分类的贡献【12】
【13】使用CNN对2-dimensional mapping of the gene expression进行癌症分类,并达到了超过95%的准确率
具体其实就是将10381个genes映射到染色体位置上,并通过补0,将每个样本都reshape成102x102的image
【14】本课题组使用一个auto-encoder system将pathways和functional gene-sets进行embedding,然后进行癌症分类
【15】本课题组还将TCGA数据随机变换成2-D data,使用CNN进行预测,也达到了95%的准确率
但gene与gene之间的调控关系(mRNA水平)还没有被用到。最近GCNs的发展,使得我们可以将这类信息融合到model中:
- 使用STRING数据库预测乳腺癌的转移【17-20】
另一个问题是去确定重要的标记物
本研究使用TCGA数据和4类gene network训练GCNs模型,对癌症进行分类,并进行了变量筛选的研究。
Methods
数据集
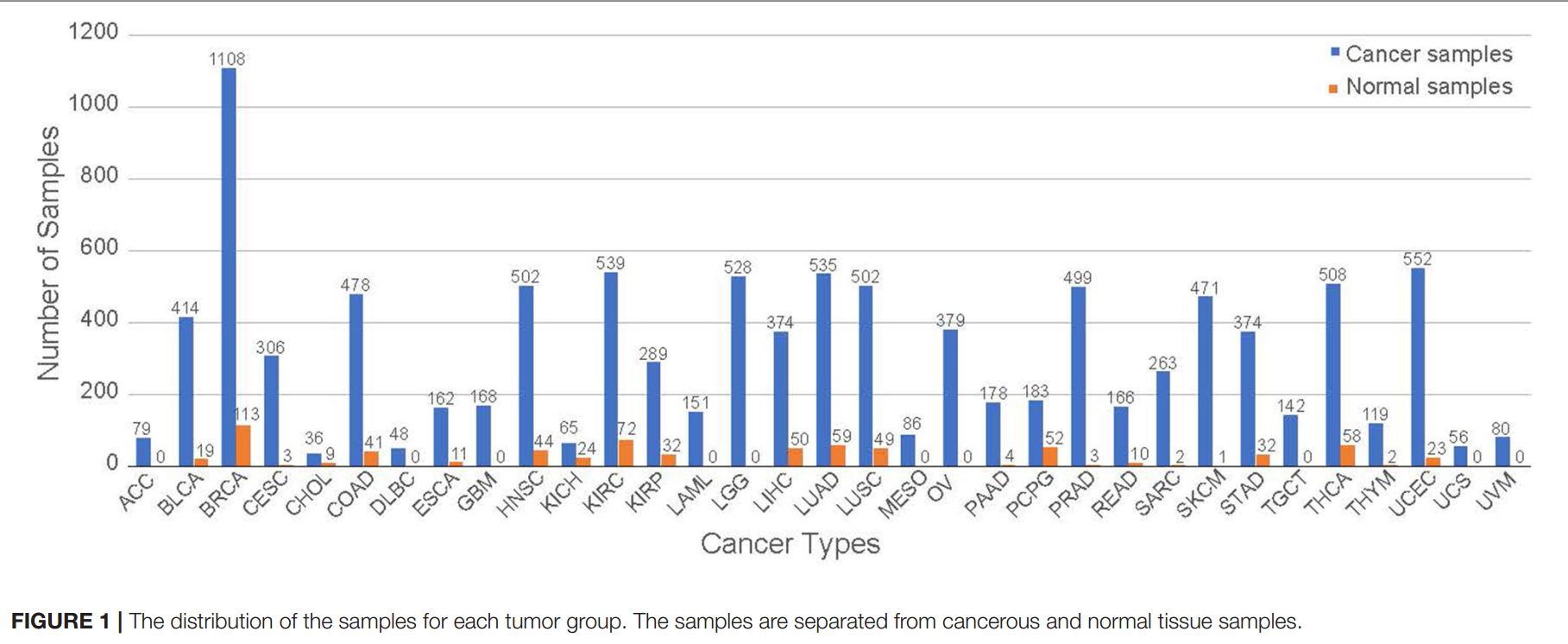
使用TCGAbiolinks【25】packages下载,共有10340个tumor samples、731个normal samples
共有56716个genes,使用指标是\(\log_2(FPKM+1)\)。
为了降低模型的复杂度,这里使用\(mean \gt 0.5\)和\(std \gt 0.8\)来进行基因筛选,得到7091个genes(拥有最多的信息)
然后将所有的表达量水平归一化到0-1之间。
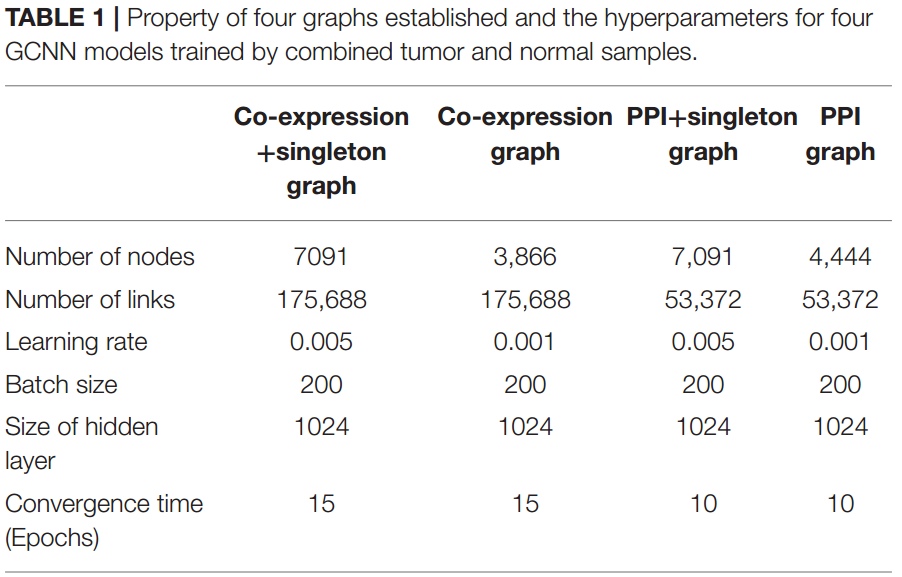
Graphs
Co-expression
使用MATLAB计算了gene间的spearman correlation【26】,然后令\(cor > 0.6\)且\(p < 0.05\)为存在一条边连接两个genes
如果将没有边相连的genes都删除,最后得到只有3866个genes之间的co-expression network,其adjacency matrix记为\(W_{co-expr}\)
PPI
来自STRING数据库【22,23】
先把7091个genes都送入BioMart databased,找到其对应的Ensembl protein IDs【27】,然后找到其STRING中的对应关系
因为非编码gene的存在,最后可以建立PPI network的genes数量是4444,记其adjacency matrix为\(W_{PPI}\)
Singleton Nodes
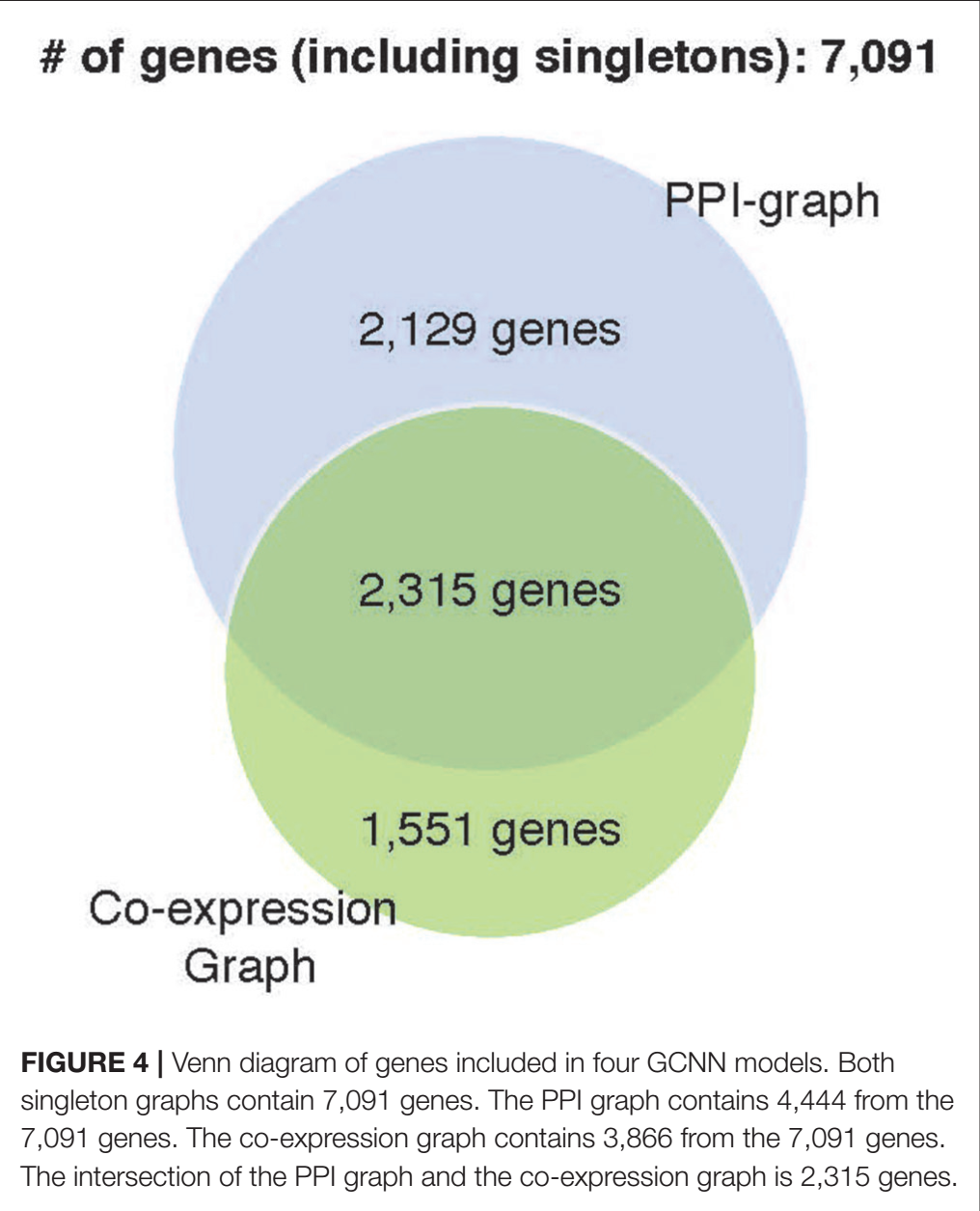
上面介绍的两个网络都没有包括所有的7091个genes。这里使用0将\(W_{co-expr}\)和\(W_{PPI}\)补充到7091x7091的维度,分别作为co-expression+singleton和PPI+singleton networks。
虽然我们使用0来进行的填补,好像这些新补充的genes并不会参与到计算中。但实际上,大多数GCNs实现时,会先将adjacency matrix的对角线补成1,或者更新节点特征的时候单独拿出一部分来考虑自身对其的影响。
singleton的补充相当于将这些节点直接加到了graph中,作为孤立点存在,补充了一部分的信息
这4个网络的基本信息如下:
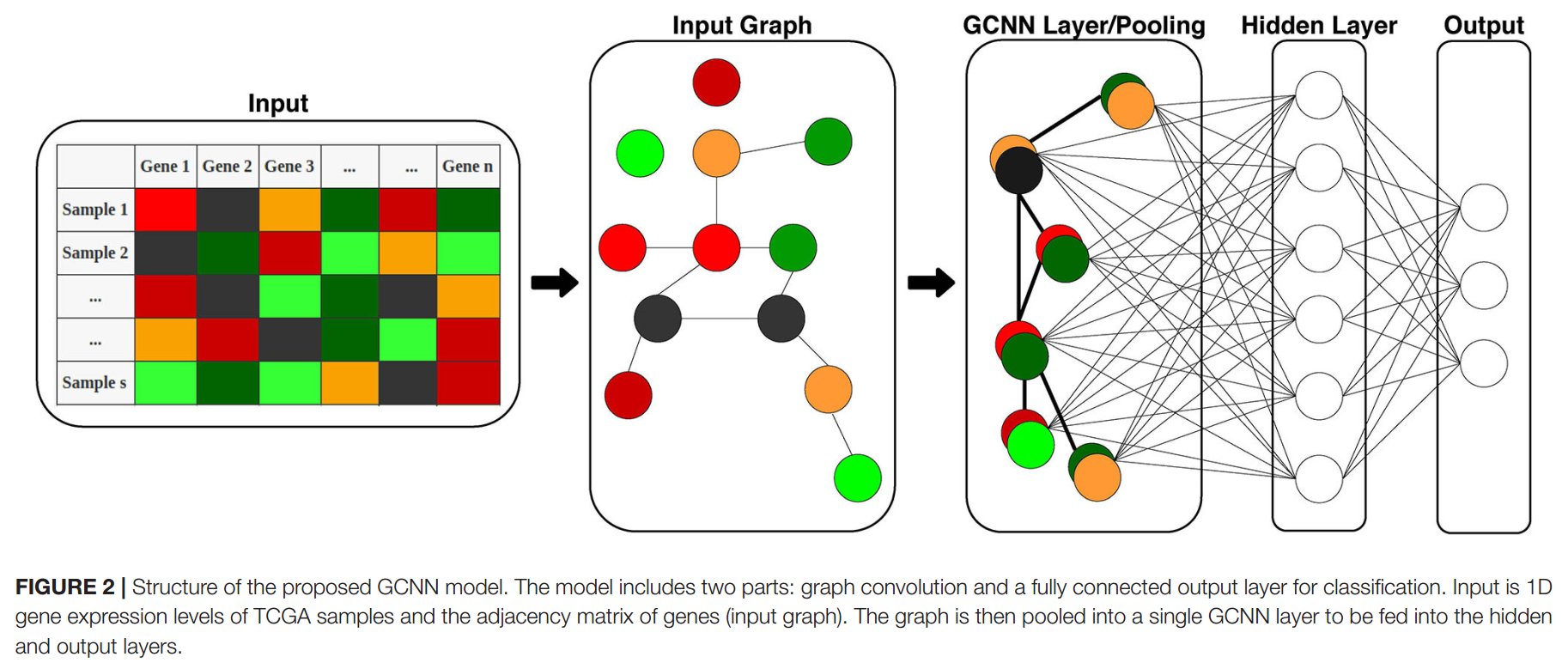
GCN models
主要包含这几部分:graph convolutional layers、coarsening or pooling、fc。
ChebConv和GCNConv:略
Coarsening (Pooling)
本研究使用一种贪婪的、简单的图粗化方法,这样每次大约降低一半的节点数量。
每次选择一个节点和其邻接点加在一起。如果选择到的是singleton node,则随机选择另外一个节点进行加和。
其使用的很有可能是ChebConv中的粗化方法。
fc和softmax
整个网络结构是一层GCN+两层coarsening+fc(1024 hiddens)+softmax。
5-CV,训练使用的是Adam,epoch=20,batch size=200。使用基于acc和loss的random search来进行超参数的调整。
这里需要进行调整的参数即学习率(0.001-0.005)。
计算gene扰动
即通过对gene添加扰动,查看扰动对预测结果的影响,来评价变量在分类任务中的重要性。【12】

筛选样本
将没有任何一类的预测概率\(\gt0.5\)的样本去掉,这些样本对于其所属的类别没有足够的代表性。
为每个分类(共34类)计算贡献分数
上面的伪代码介绍的更加详细
对于一个样本,将某个gene的值改为0和1,重新计算在该样本的类上的概率,记录变化最大的那个值为score。将某类的所有样本的score都加和在一起,即该gene对该类的贡献分数。
最终我们得到的贡献分数是一个分类数(34)x genes数量的一个matrix。
Normalization
对于每一类别,将上面得到的贡献得分归一化到0-1之间。
如果想,还可以将所有的cancer的列别的归一化贡献得分加总,得到一个Overall Cancer的得分。
Results
预测准确性
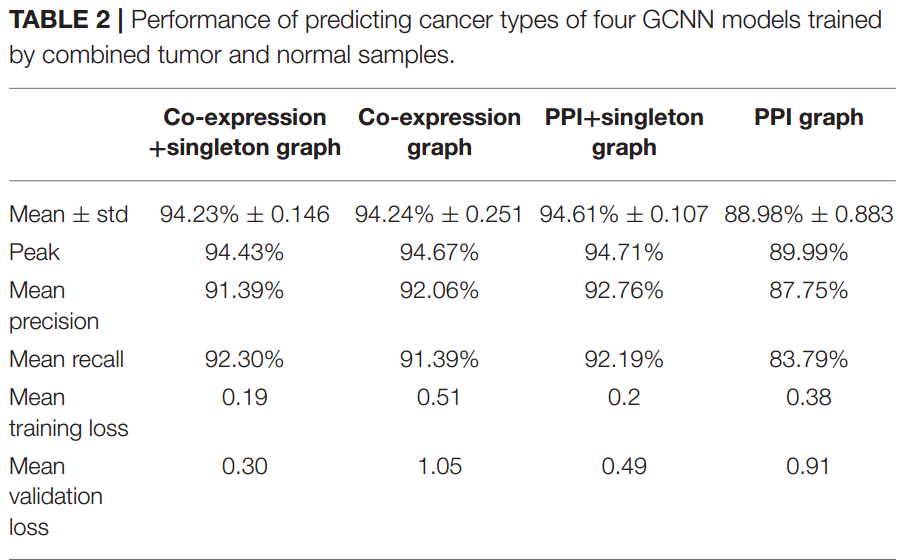
上图是预测结果。
PPI model的预测准确性是最低的,其只含有可编码为蛋白质的gene和基于STRING数据库的交互作用。非编码gene没有包括其中,许多其他类型的gene调控也没有被包含其中,这可能导致了其性能较差。
PPI+singleton model有超过5%的性能提升,提示没有被包括在PPI中的gene可能也起到了重要的作用。
co-expression model拥有类似PPI+singleton model的性能,增加singleton并没有得到明显的提升。这可能是因为我们co-expression networks中已经包括了一些重要的非编码genes。从另一个方面说,这也说明剩下的这些singleton genes是“不太重要的”,其本身也是没有通过相关性检验的。
在附录中有5-CV训练过程中的loss和acc的收敛情况,用来判断是否存在过拟合。其中使用PPI网络的model拥有最长的收敛时间但最低的validation loss。
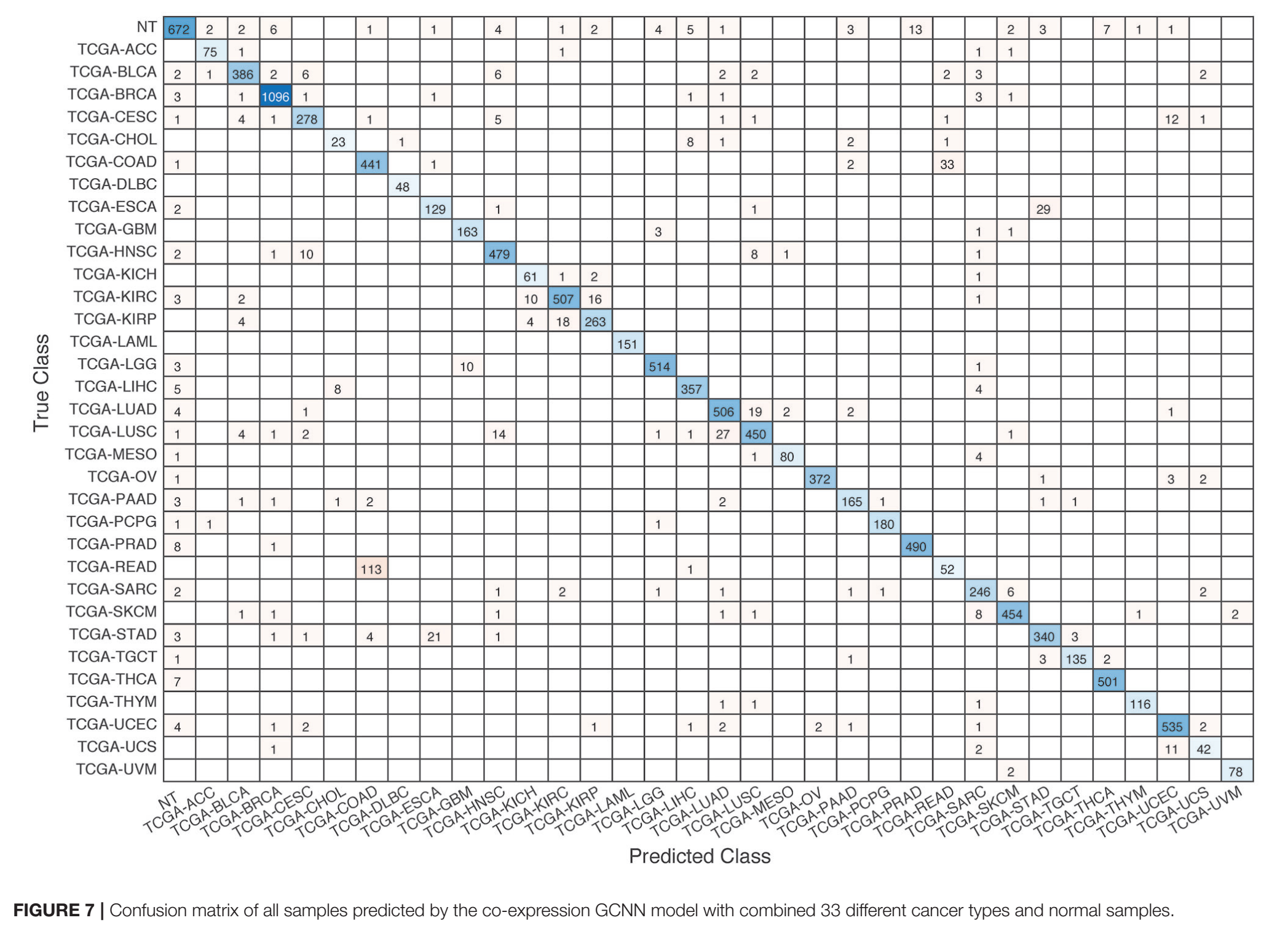
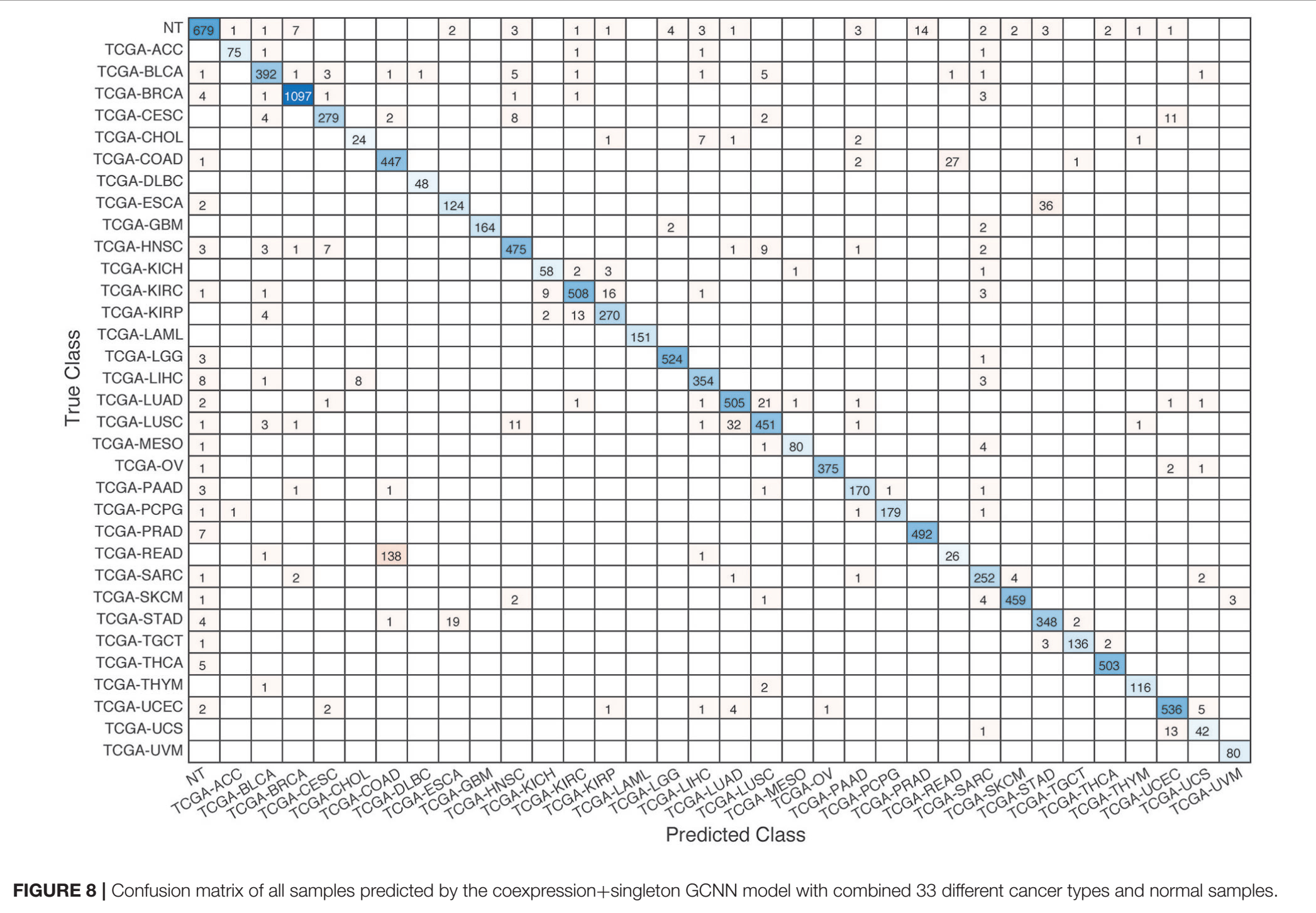
进一步对co-expression和co-expression+singleton models的结果进行precision-recall分析和confusion matrix分析,发现:
- READ(直肠癌)和COAD(结肠癌)的错分是导致性能降低的关键所在。另外错分比较多的还有CHOL(胆管癌)和LIHC(肝细胞癌)、UCS(子宫癌肉瘤)和UCEC(子宫内膜样癌)。
- 以上这些错分都存在两个特点:错分的pair基本都在同一个器官或组织中,每一对pair中都存在一类其样本量非常少
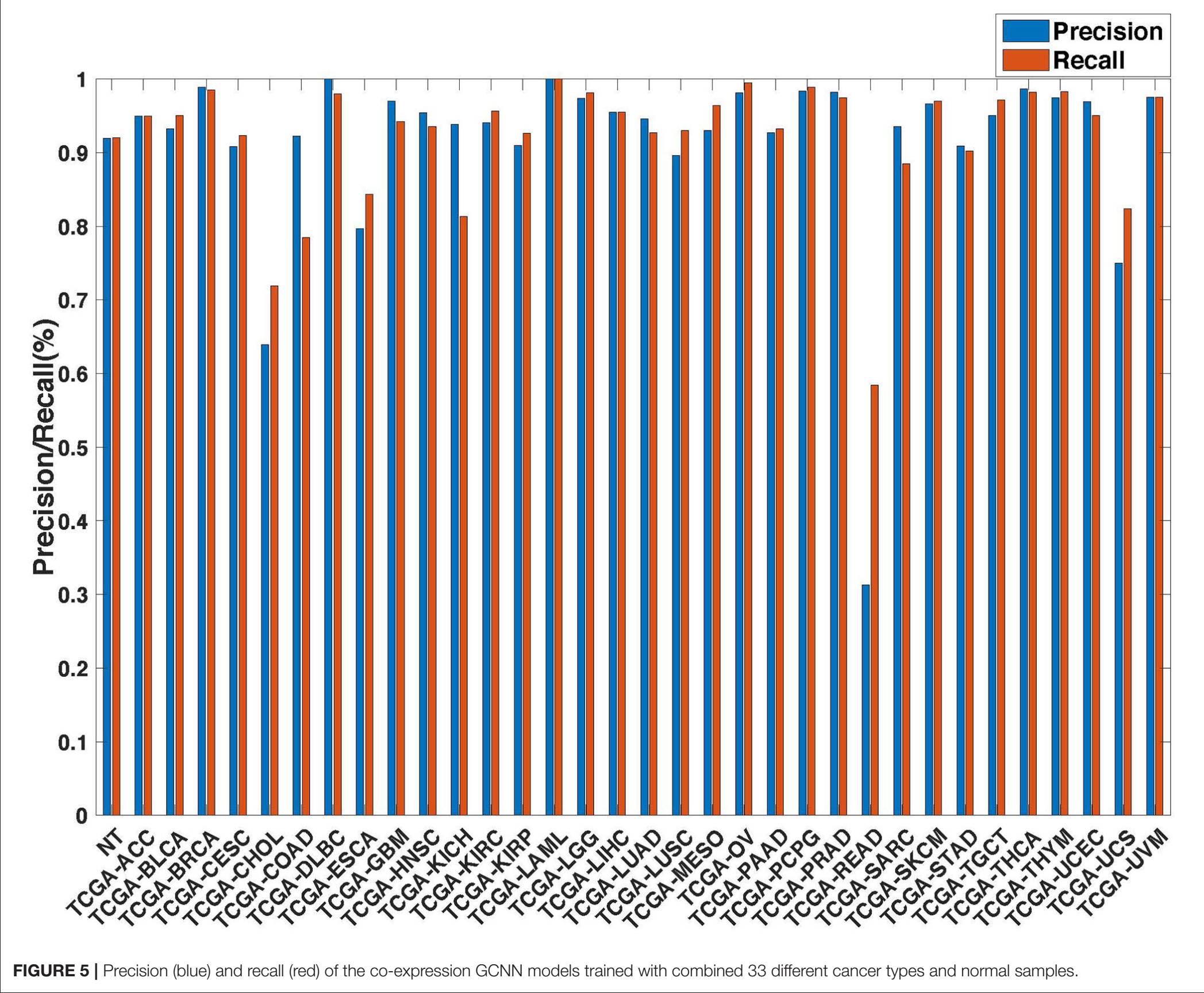
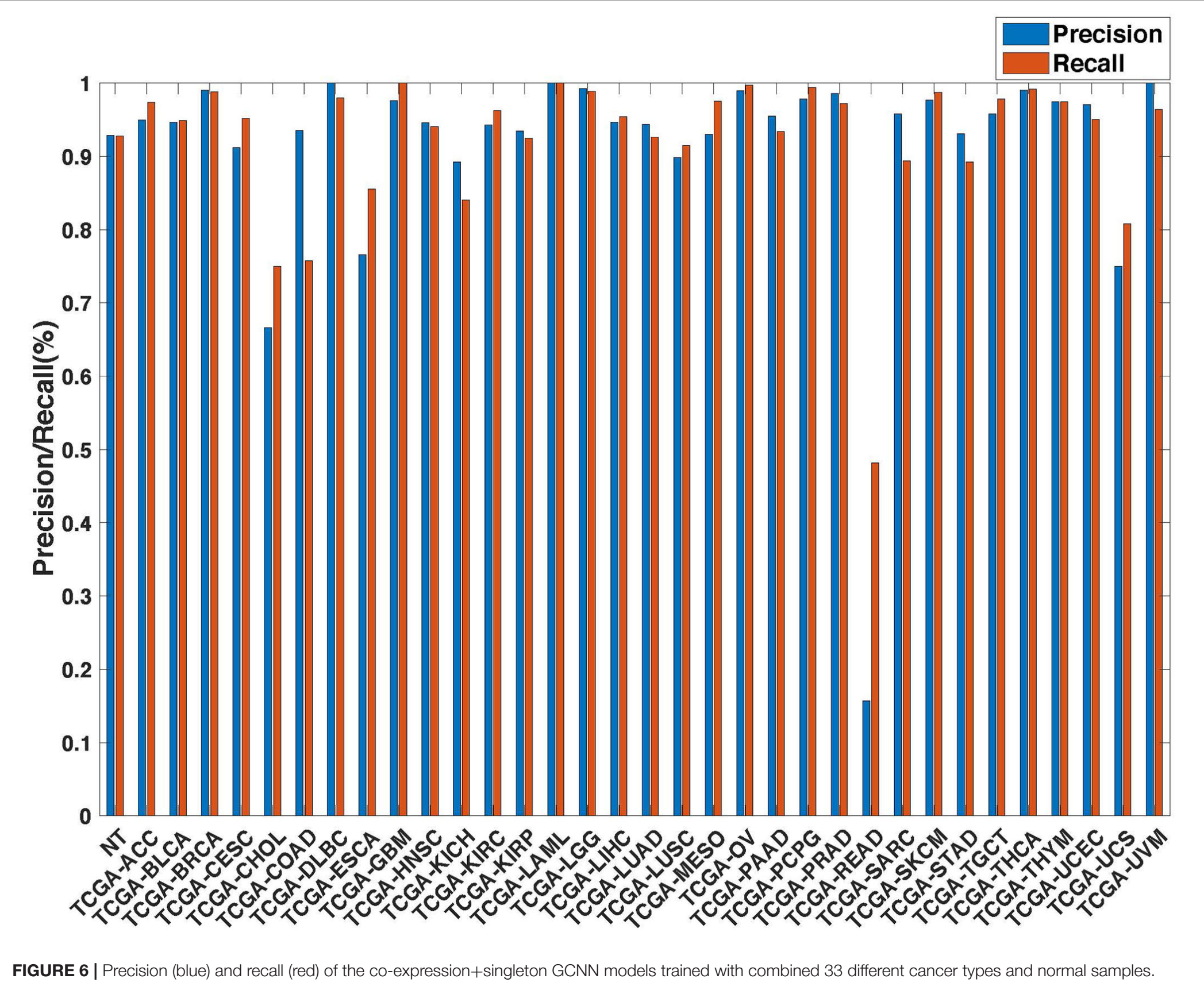
癌症特异性
因为癌症类型不同一般也意味着组织类型不同,所以我们建立的分类模型可能只是捕获到了其分类组织的信息(即tissue-specific)。这显然不是我们想要的,所以这里是来讨来model捕获到的信息除了tissue-specific,还有cancer-specific。
之前的研究都进行癌症的分类,而且效果都不错【32,10,13】。但这些研究都没有包括normal samples,所以无法判断其分类是tissue-specific还是cancer-specific。最近的研究【12,15】提示可能是cancer-specific的。
本研究去看了一下normal samples的分类结果:92%,也就是731个中的672个样本都被分对了,无论这些样本来自哪些组织器官,其都被正确分类,说明model使用的更多的是cancer-specific信息。
建模后分析
gene perturbation的方法进行gene筛选,会让那些孤立点更容易被选择,所以这里不使用加了singleton的graph model。又因为PPI graph model的性能比较差,所以这里选择co-expression graph model进行建模后分析。
非孤立点被扰动后,其信息可以有其邻接点信息进行补充。所以其带来的性能的差异可能不会太大,就不容易被选出。
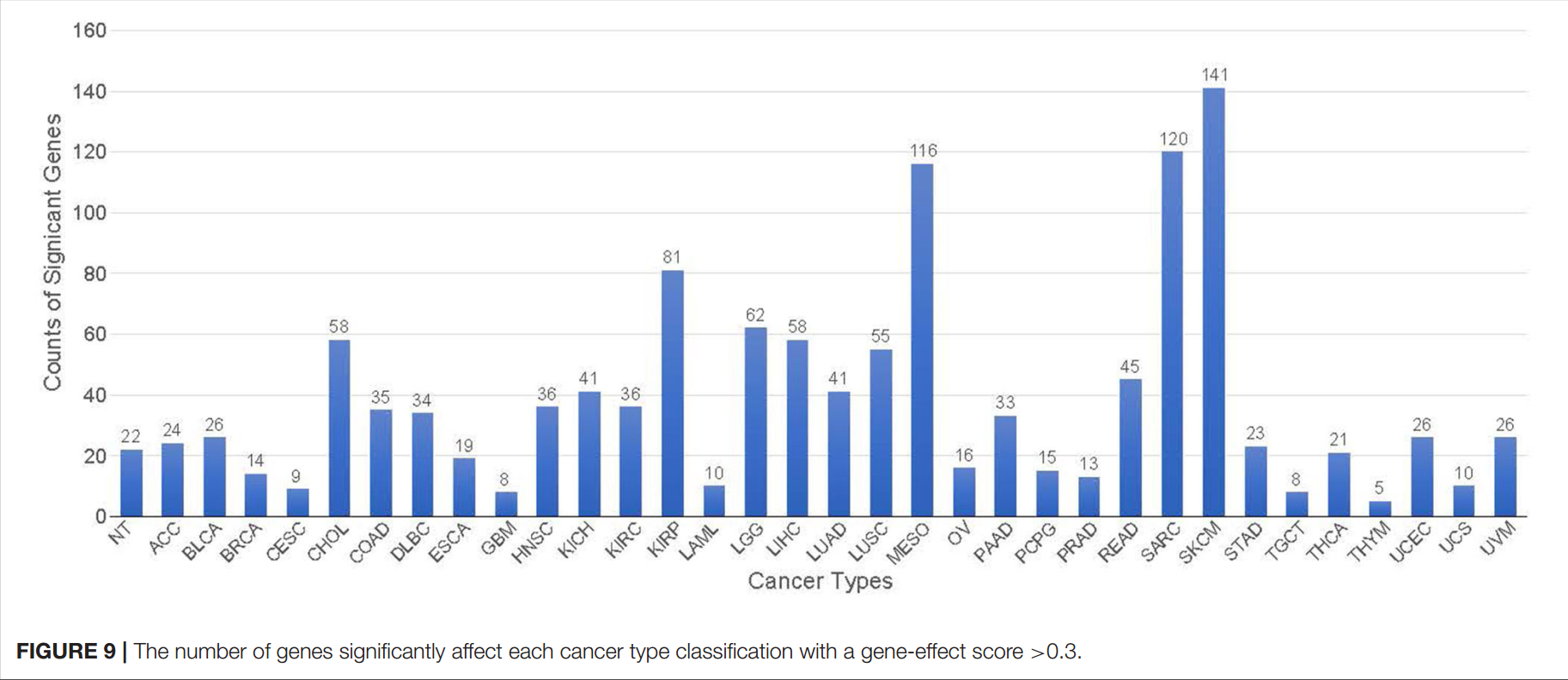
使用0.3的阈值(查看直方图选择的),我们一共找到了428个潜在的biomarks。这些biomarks在每类癌症上的分布:
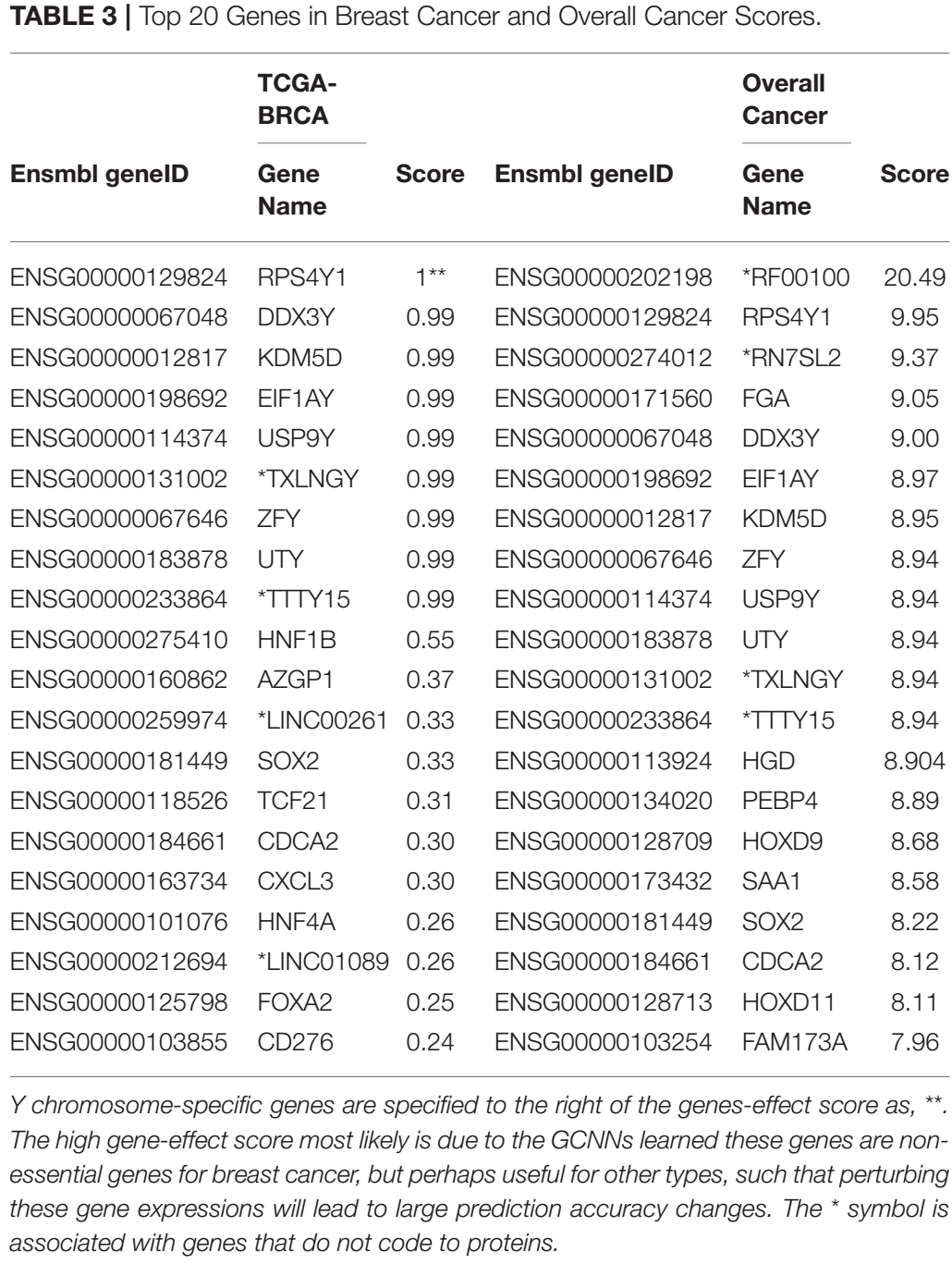
以breast cancer为例,其排名前20的genes中,前9个gene都是Y染色体相关的,即其学习到了判断乳腺癌的第一个关键是性别。剩下的genes也在breast cancer的研究中报道过。
Discussion
本研究使用的genes筛选的标准(mean > 0.5和std > 0.8)是否可靠,GCN model对于此阈值选择的敏感性需要进一步的研究。
co-expression graphs建立时使用的阈值(cor > 0.6和 p < 0.05)是最好的那个,如何确定这个阈值可能需要进一步的研究。
除了FPKM外,还有TPM。本研究从UCSC TumorMap上下载了TPM数据,并将其和FPKM数据进行了比较。发现不管数值上的、还是依据其进行的co-expression都是非常相似的。所以两者应该会有相似的结果。
MI可以捕获到非线性信息,但其计算复杂。
在之前的研究中【36,37】,比较了MI-based和correlation-based methods之间的表现和效率,correlation-based method表现更好。
很多研究【38】也认为,gene间的关系大多是线性的、单调的,所以本研究选择使用correlation。
我们可以将PPI和其他的gene关系(如调控等)综合成一个完整的graph,进行相关研究。
一个可行的方法是进行文献数据挖掘,比如【39】。
深度学习是纯粹数据驱动的,而如果想要将生物学信息融入其中,可能需要对GNNs进行彻底的修改来完成【40,41】。



