Deep Learning Based Tumor Type Classification Using Gene Expression Data
- 杂志: bioRxiv
- IF: None
- 分区: None
Introduction
Next Generation Sequencing(NGS)技术的发展极大地提高了人类基因组学的研究,其中TCGA计划是其中的重要代表。
利用这些数据一个目的是筛选biomarkers,但这一般都是data specific的,无法利用到其他癌种的信息。
另外,利用所有的癌种数据对癌种分类建模本身有着其诊断的意义。但在这个任务上也有着困难:
变量(genes)多,很多genes是weak features,在许多模型中会遇到“维度灾难”的问题;
虽然深度学习在基因组学还处于开始阶段,但其在计算机视觉上的成功毋庸置疑。另外,如Taylor decomposition、layer-wise decomposition和Grad-CAM【16】等可视化方法也增强了深度学习的可解释性。
这提示我们可以利用深度学习技术来建模癌种分类模型,其适合进行变量多、关系复杂的建模。
更进一步,如果我们对癌种分类贡献较大的变量更加重要,则我们可以利用上刚才我们提到的可视化方法得到变量的重要性评分,筛选biomarkers。
本研究首先使用方差来筛选变量,然后将高维表达数据(\(10381\times1\))整理成2-D image(\(102\times120\)),然后使用3-layers的CNN对其进行分类,并使用10-CV对其进行评价。基于Guide Grad-CAM【16】,我们对每个类生成重要性热图,得到biomarkers,并在功能分析中证明了筛选的biomarkers都是有生物学意义的。
相关工作
GA/KNN【11】方法,进行多癌种分类,在31类上得到了90%的正确率,并且可以生成一个top genes set。
【5】首先使用stacked auto-encoder来提取high-level features,然后进行ANN中进行分类,其在癌或非癌的分类任务上得到了94%的acc。此模型如果应用到多癌种分类上,将会变得特别复杂,难以实施。
为了得到top set genes,其将stAE中每层的权重相乘从而得到每个gene的权重。这个想法和本研究中使用的可视化方法类似。
Deep Taylor decomposition、layer-wise relevance propagation(LRP)【1,2】和Guided Grad-CAM是在深度学习领域进行解释性可视化的方法,本研究使用的是Guided Grad-CAM。
Methods
流程图如下:
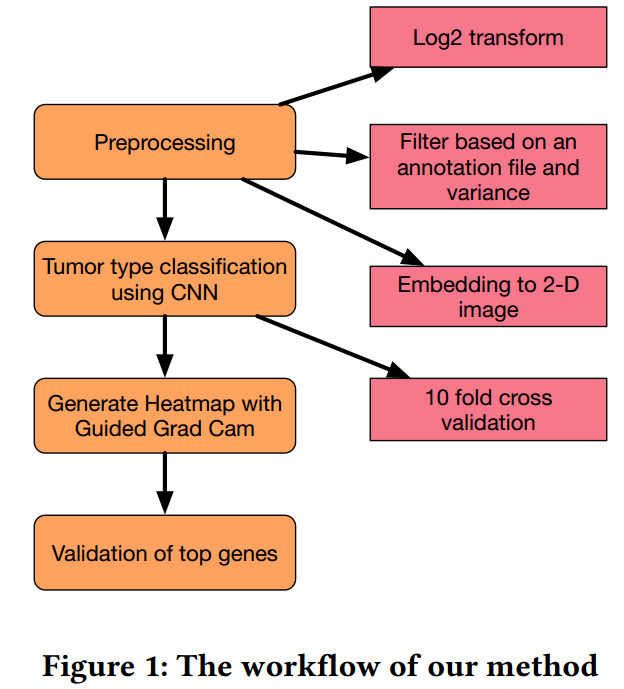
数据
共有33个癌种,10267个样本,20531个genes。
预处理
- 数据是normalized read count,首先将小于1的数值变成0,并进行\(log_2(x+1)\)变换。
- 将genes match到annotation file(04/03/2018,from NCBI),其中有1000个genes没有在annotation file中找到了,并使用var=1.19来进行变量筛选,最终得到10381个genes。
- 首先根据其染色体编号,将其排序,然后将其reshape成\(102\times102\)的image,在最后一行pad zeros来加成。
- 将所有生成的imagesnormalized到0-255范围内。
最后生成的figures:

分类
使用的架构如下图所示:
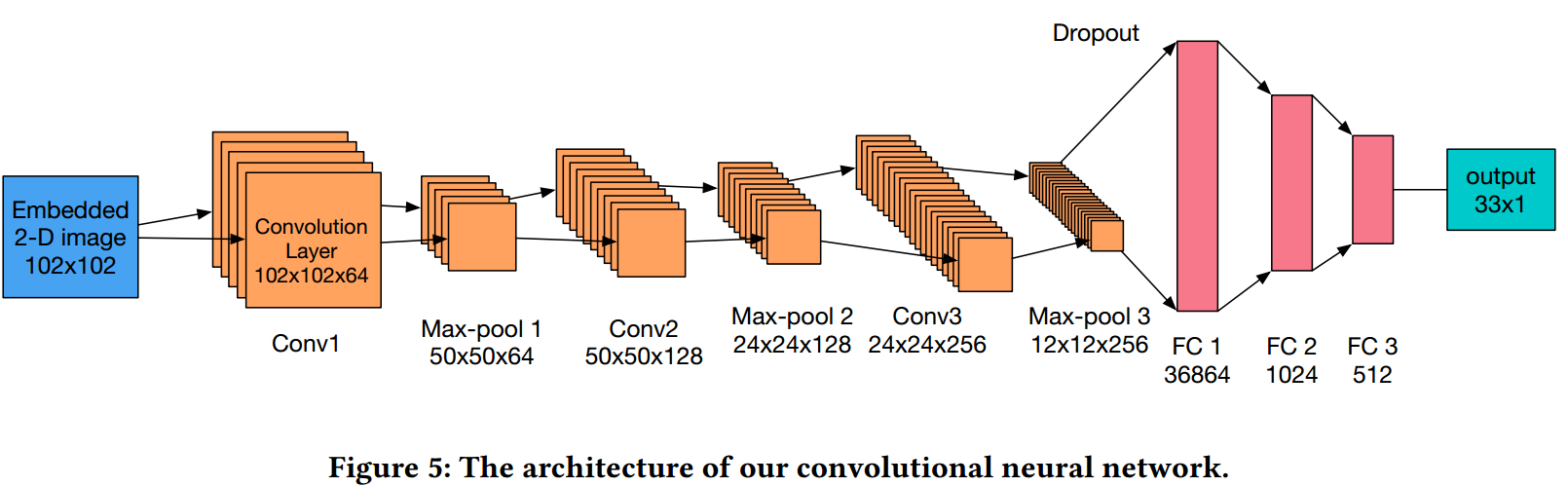
在进入fc之前加了一个0.25的dropout,进行了10-CV来进行评价。
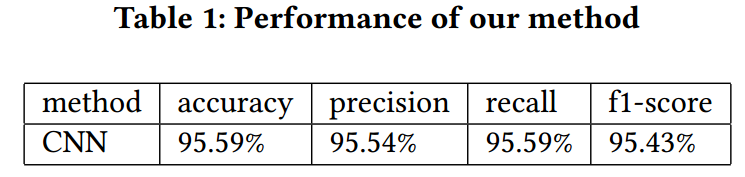
使用的评价指标有ACC、average precision、average recall、average F1 score。
热图
利用guide Grad-CAM可以为每一张image的每一绘制heat-map,其中值越高代表该pixel在最后的分类中贡献更大。
验证
- 每个癌种选择top 400 genes来进行pathway analysis。
- 研究前一步中没有人格结果的tumor type的top 5 genes,来寻找其和肿瘤的关系。
Results
分类
结果如下:
进一步,我们绘制了confusion matrix:
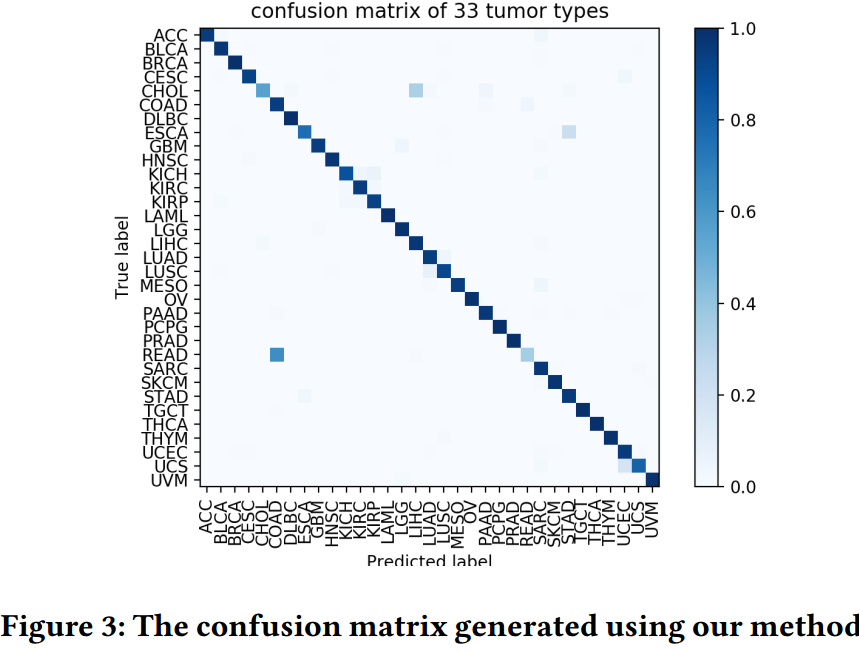
可以看到:
- READ(直肠癌) samples容易被错分为COAD(结肠癌),这可能是因为两类癌症有着比较近的空间关系。
- 许多CHOL(胆管癌)样本会被错分为LIHC(肝细胞癌),这可能因为CHOL的样本量太少。
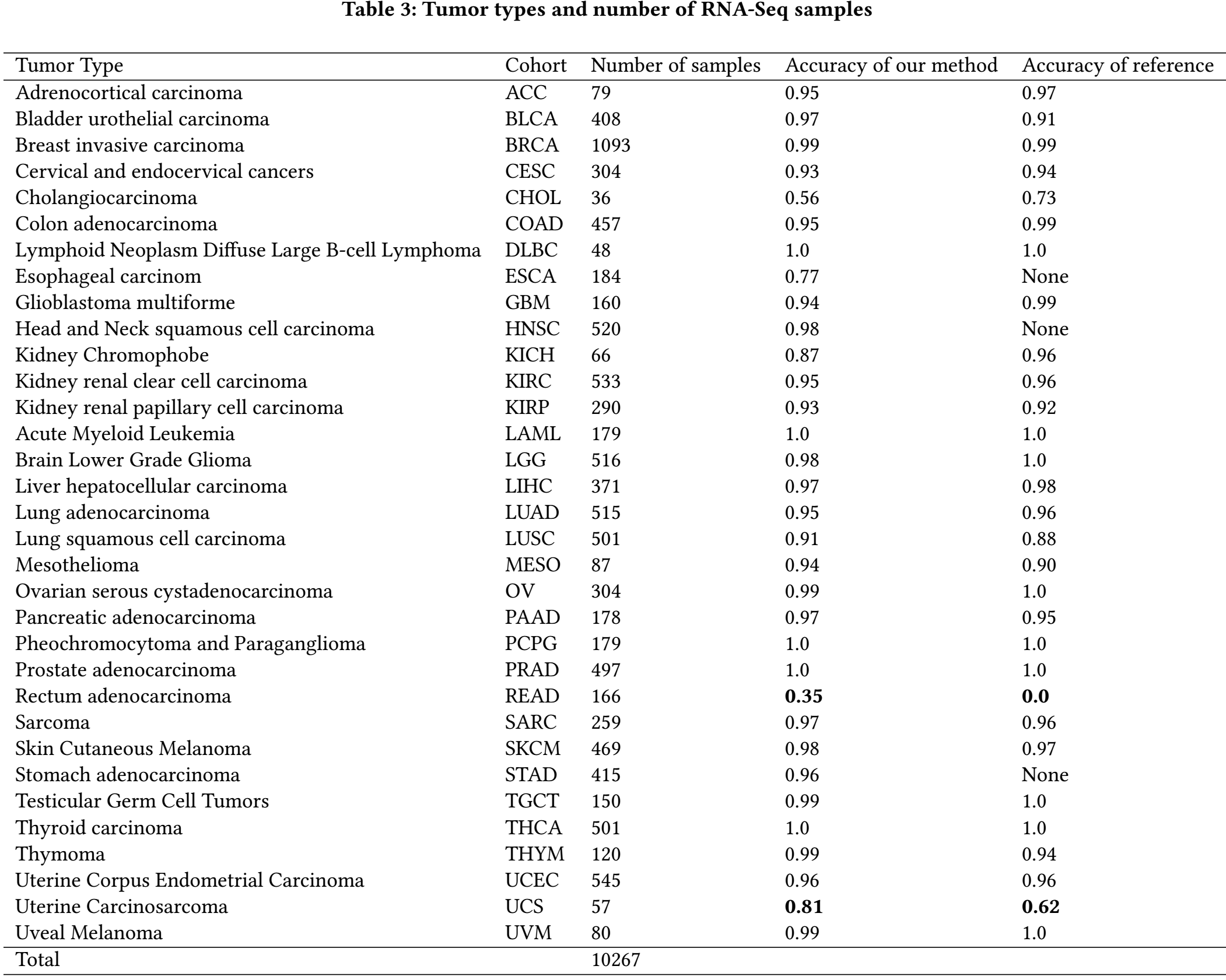
tab3进一步显示了在每个癌症类型上和【11】的比较,可以看到在READ和UCS(子宫癌)的分类上,本研究的方法有着较大的提升。
生成的热图

上面的每一行表示一种癌症,而每一列表示一折的结果。从上面的例子中我们可以看到,尽管每一折产生的热图有所不同,但还是显示出清晰的、相似的模式。
Top genes的验证
对于每一类的热图,我们将其intensity的变化绘制成fig4:
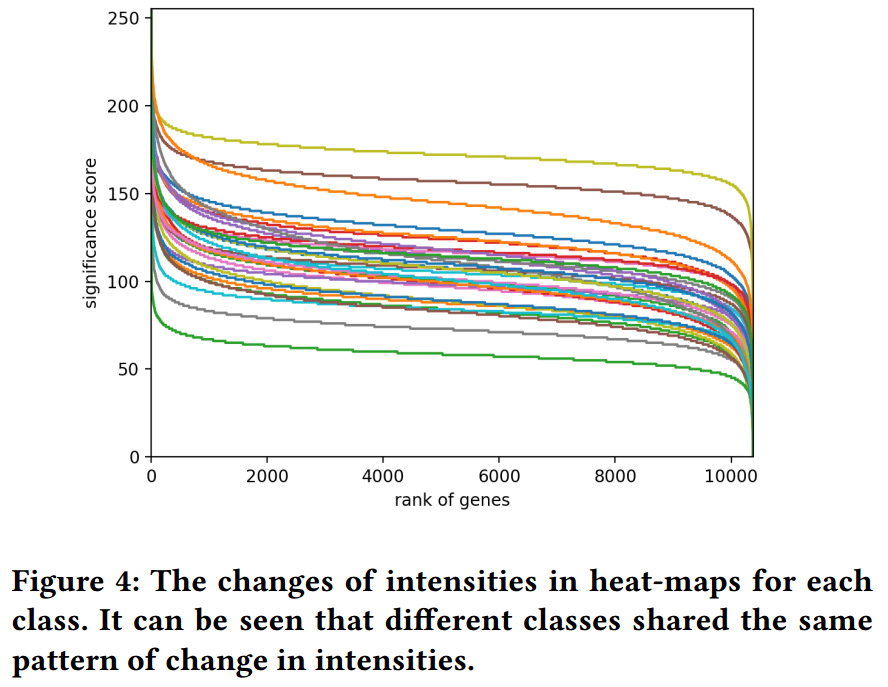
我们可以看到每一类都表现出相似的性质,即在top 100 genes上其scores下降的很快,而从大约400个genes开始,其下降开始变得非常平缓,最后又有一个非常迅速的下降。这说明top 400 genes可能拥有更强的作用,所以选择这些genes进行富集分析。
使用David website来进行KEGG pathway analysis,其中p值小于0.001的展示在tab4中:
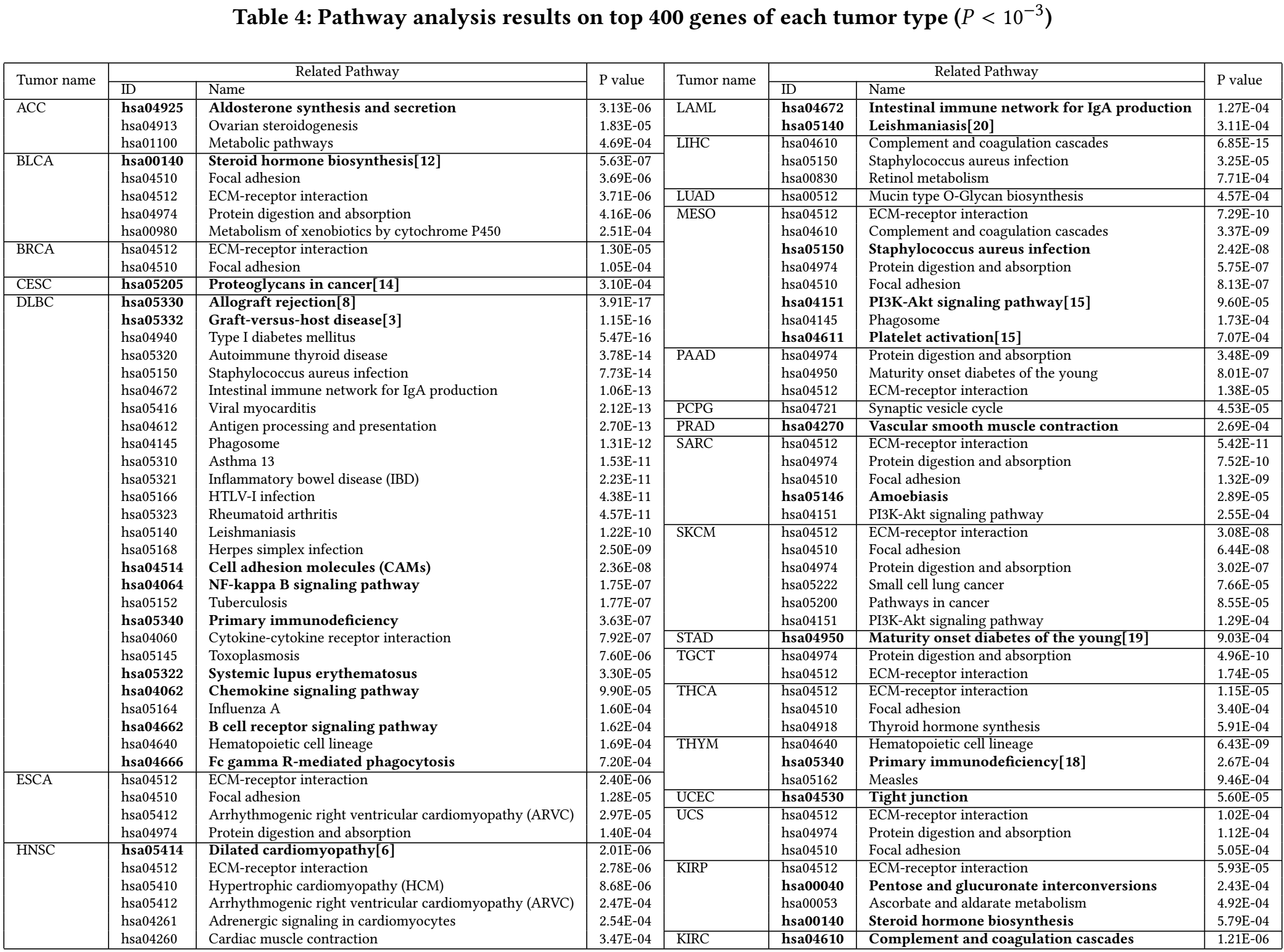
其中有24个癌种被发现有显著的通路富集,其中16个癌症在这400个genes中有至少一个相关通路,在这些通路中的genes可以看做是biomarkers。
另外8个癌种中,虽然没有明确的相关通路,但有两条显著的富集通路:hsa04512和hsa04510,但其在不同癌种中的genes不相同。
在剩下的9个癌种中,没有显著的通路。因为CHOL和KICH的样本量有限,我们忽略他们。READ的预测acc很低,我们也忽略。然后我们查看他们的top 5 genes,并在GeneCards中查看他们的信息:
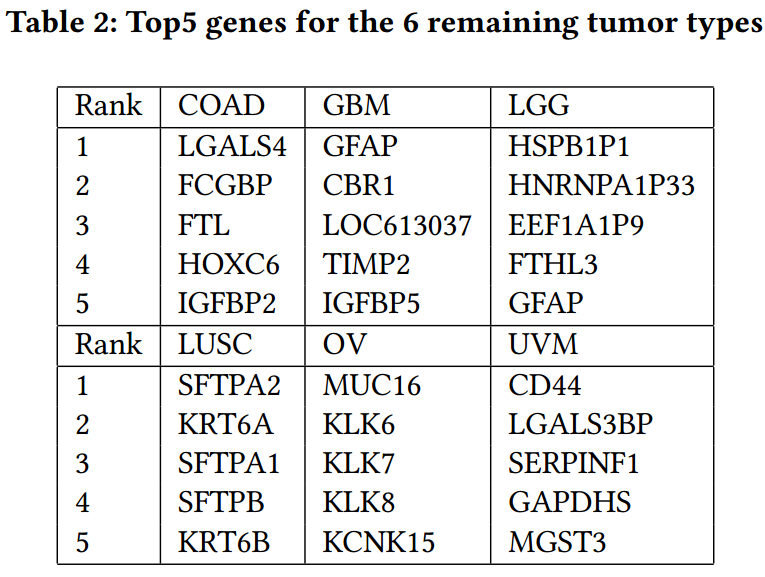
OV中的MUC16就是CA125,被认为是OV中唯一可靠的诊断标志物。
可以通过查看这些genes的信息,确定筛选到的genes中有一部分和癌症密切相关,可以看做是潜在的biomarkers。
Discussion
通过错分样本的分析,限制本研究模型性能的一个原因可以在于imbalanced dataset,所以使用一些oversampling methods(SMOTE)可能有助于性能的提升。



