GSAE: an autoencoder with embedded gene-set nodes for genomics functional characterization
- 杂志: None
- IF: None
- 分区: None
Introduction
gene set分析来解释gene expression data,是一个非常重要的生物信息学工具。
首先是常用的功能富集,比如常用的GSEA【1】、DAVID【2,3】和其他【4-6】。
另外还可以用来进行分类,比如PAM50【7】和【8,9】等。
但也有一些研究指出,不同研究所得到的gene set之间重合度较小【10】,所以最好通过整合多个不同的gene sets来提高其一致性。
最近深度学习的发展开始渗入到分子和细胞表达数据的分析中。
CNNs用来进行DNA-protein结合位点的预测【11】或用来检测表型相关细胞亚型【12】。
【13-15】使用AEs来进行降维。
【13】提出了一种NN model,其整合PPI和PDI(protein DNA interaction)了对single-cell RNAseq数据建模,但其只是使用这些先验信息来提高降维和细胞特异性识别的性能,而没有分析合并PPI节点所带来的影响。
本研究中提出Gene Superset AutoEncoder(GSAE)模型,其利用到了一个先验定义的gene sets来保证一些生物学特性。
本研究提出了gene superset的概念,目的是来确定学习到的gene supersets间的功能性或临床上的相关性。
使用TCGA的数据进行了验证。
Methods
数据集
来自TumorMap【16】整理的TCGA Pan-cancer RNAseq dataset,有9806 samples,33 cancer types。
使用BRCA(1099 samples)来characterize network nodes。
使用LUAD(515 samples)来进行survival analysis。
使用LUAD、BRCA、LGG(523 samples)和SKCM(469 samples)来比较supersets的复现性。
使用的表达量数据是TPM,并进行了log transform(\(\log_2(TPM+1)\))。
Gene superset autoencoder
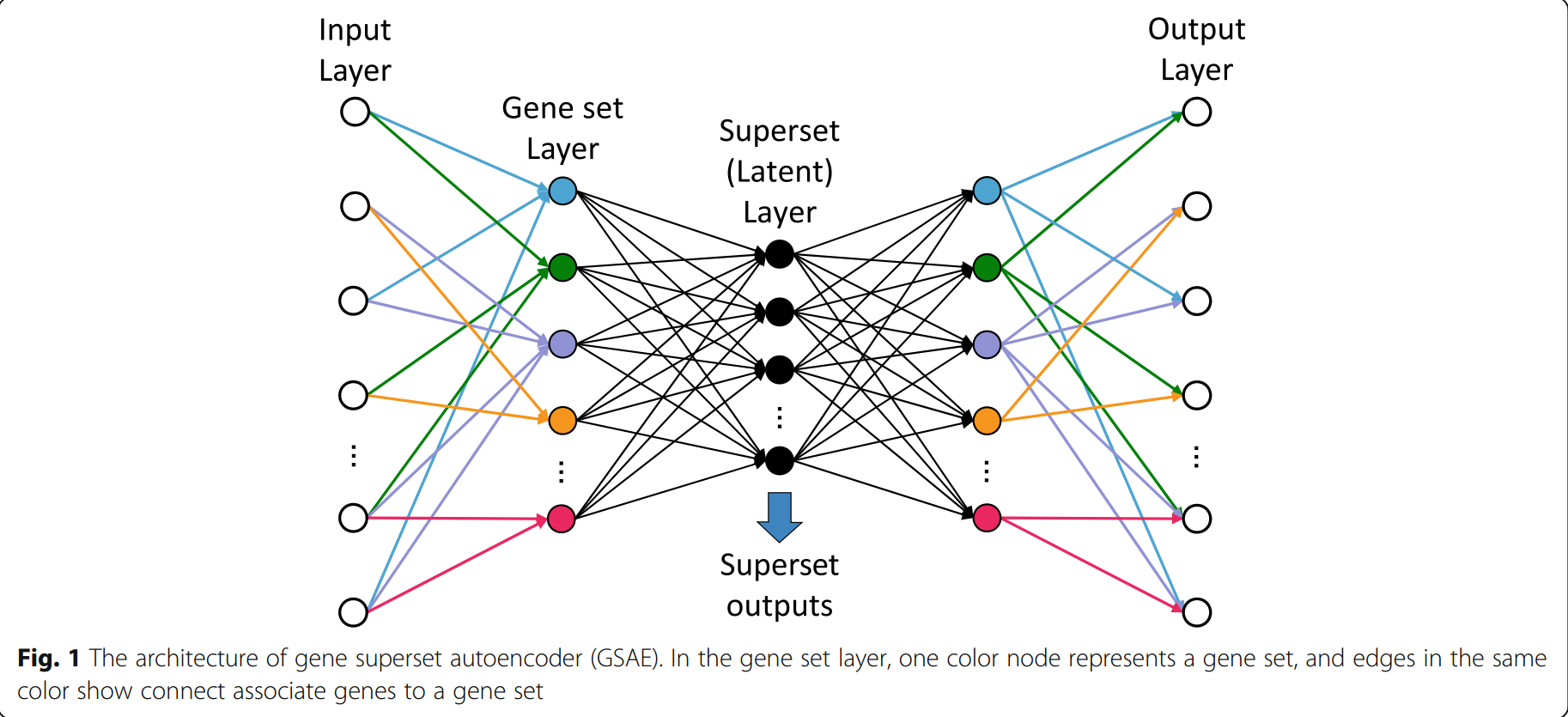
GSAE的结果如上图所示。
就是一个AEs。
将基因集纳入编码层
将encoder的第一个hidden layer的节点作为gene set的表示,具体做法就是:表示某一具体gene set的节点只和在这个gene set中的genes连接。【13】使用的数据集是来自MSigDB【1,17】的CGP collection。
然后将gene set layer的输出输入到fc layer中,得到是superset表示(即AEs的瓶颈层表示)。这里的每个节点被看做是一个superset,是gene sets的组合表示。这里superset layer的节点数目是200。
消除基因集间的依赖性
CGP collection中的gene sets很多拥有高度的相似性,所以需要降低其依赖性。
这里使用的方法是【18】中的方法:
首先忽略小于15或大于500个genes的gene sets;
这也是GSEA的设置。
计算gene sets间的kappa统计量,然后将\(p\lt10^{-7}\)的gene sets进行聚类,使用最大的那个gene sets作为这个cluster的表示
最后,我们得到了2334个CGP sets和18107个genes。
实现
使用Keras 1.2.2。参数初始化使用的是uniform initialization。训练使用的optimizer是SGD(\(lr=0.05,decay=10^{-6},momentum=0.9,Nesterov=1\)。
使用5%的数据作为validation,并使用early stopping防止过拟合。
其他方法
- t-SNE,用来将supersets结果降维到2。
- HDBSCAN,用来将t-SNE的结果进行聚类。
聚类结果的评价
Dunn index:
\[\frac{min_{i,j,i\ne j}d_B(C_i,C_j)}{max_kd_W(C_k)}\]
其中\(d_B(C_i,C_j)\)表示cluster \(C_i\)和\(C_j\)间的类间距,\(d_W(C_k)\)表示cluster \(C_k\)的类内距离。
R package clv
Silouette index:在所有clusters上的mean silhouettes的mean。
R package clValid
inter-intra distance (IID) index:
\[\frac{\frac{1}{n_B}\sum_{i,j;i\ne j}d_B(C_i,C_j)}{\frac{1}{n_W}\sum_kd_W(C_k)}\]
其中\(n_B\)和\(n_W\)表示cluster pairs和clusters的数量。
差异superset分析
使用HDBSCAN对t-SNE的结果分亚型,然后使用单尾的Mann-Whitney-Wilcoxon U test来寻找两组间的差异supersets。其中在group1中值更大的称为up-supersets,反之称为down-supersets。
然后我们构建第\(i\)个gene set对第\(j\)个superset的贡献,即gsScore:
\[gsScore_{ij}=(\mu_1^i-\mu_2^i)\times w_{ij}\]
其中\(\mu_1,\mu_2\)分别表示第\(i\)个gene set在两组的均值,\(w_{ij}\)是model中第\(i\)个gene set与第\(j\)个superset间连接的权重大小。
在up-supersets中,gsScore大于positive cutoff的gene sets被选择;而在down-supersets中,gsScore小于negative cutoff的被选择。
sueprset上的生存分析
对于每个superset和gene set,使用median分为两组,然后比较两组的生存差异。
Results
在低维编码中依然保留了癌症类型的信息
这里使用TCGA PanCancer RNAseq logTPM数据,使用\(\mu\gt1\)和\(\sigma\gt0.5\)来筛选到15975个genes,9806个samples,然后使用GSAE提取supersets features。在supersets features上使用t-SNE,并可视化:
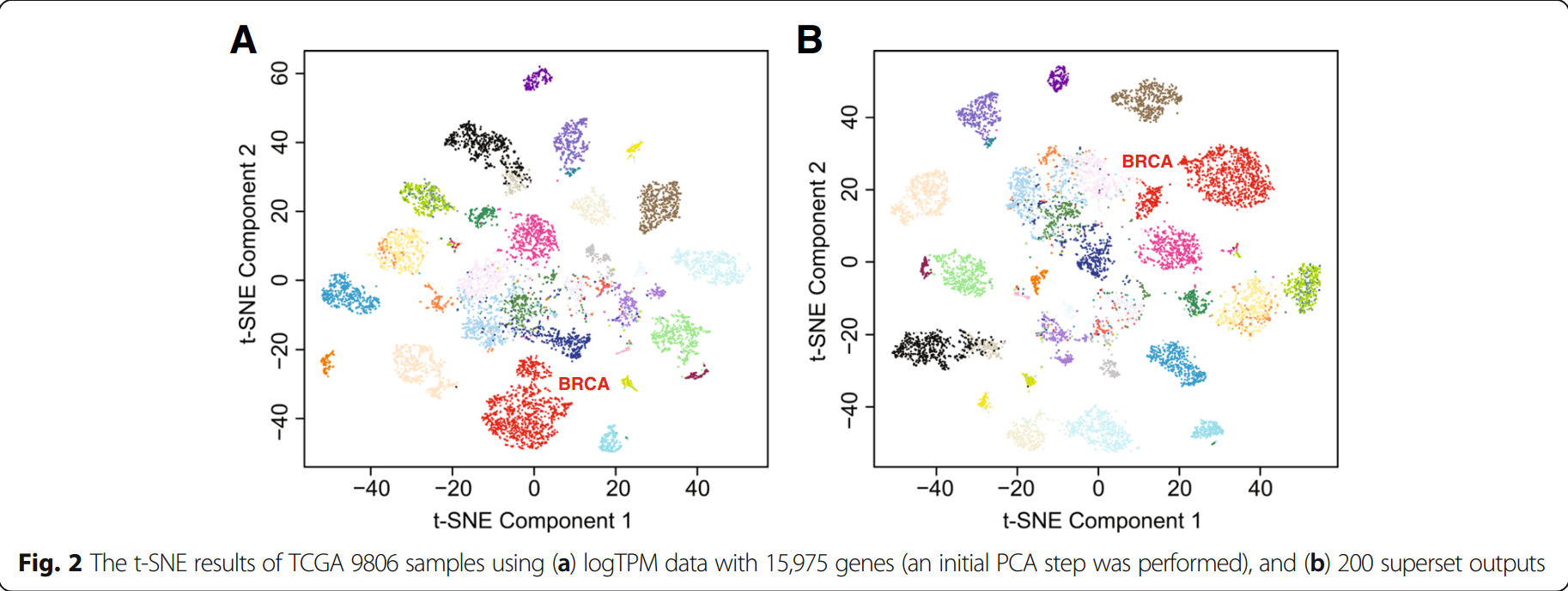
我们可以看到,使用GSAE并没有导致癌症分类信息的丢失。进一步用3种聚类评价指标来衡量GSAE降维前后的相似性:
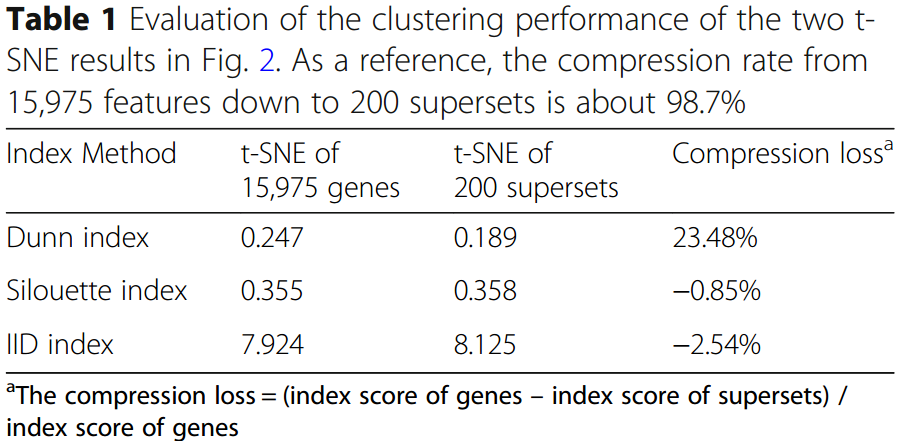
尽管前后维度降低了接近98%,但综合上面的3个指标的结果可以得出结论:GSAE在降维的时候并没有导致癌症类型相关信息的丢失。
与乳腺癌亚型相关的gene sets
上面fig2中可以看到,BRCA被分成了2个部分,我们对此进行进一步的研究。
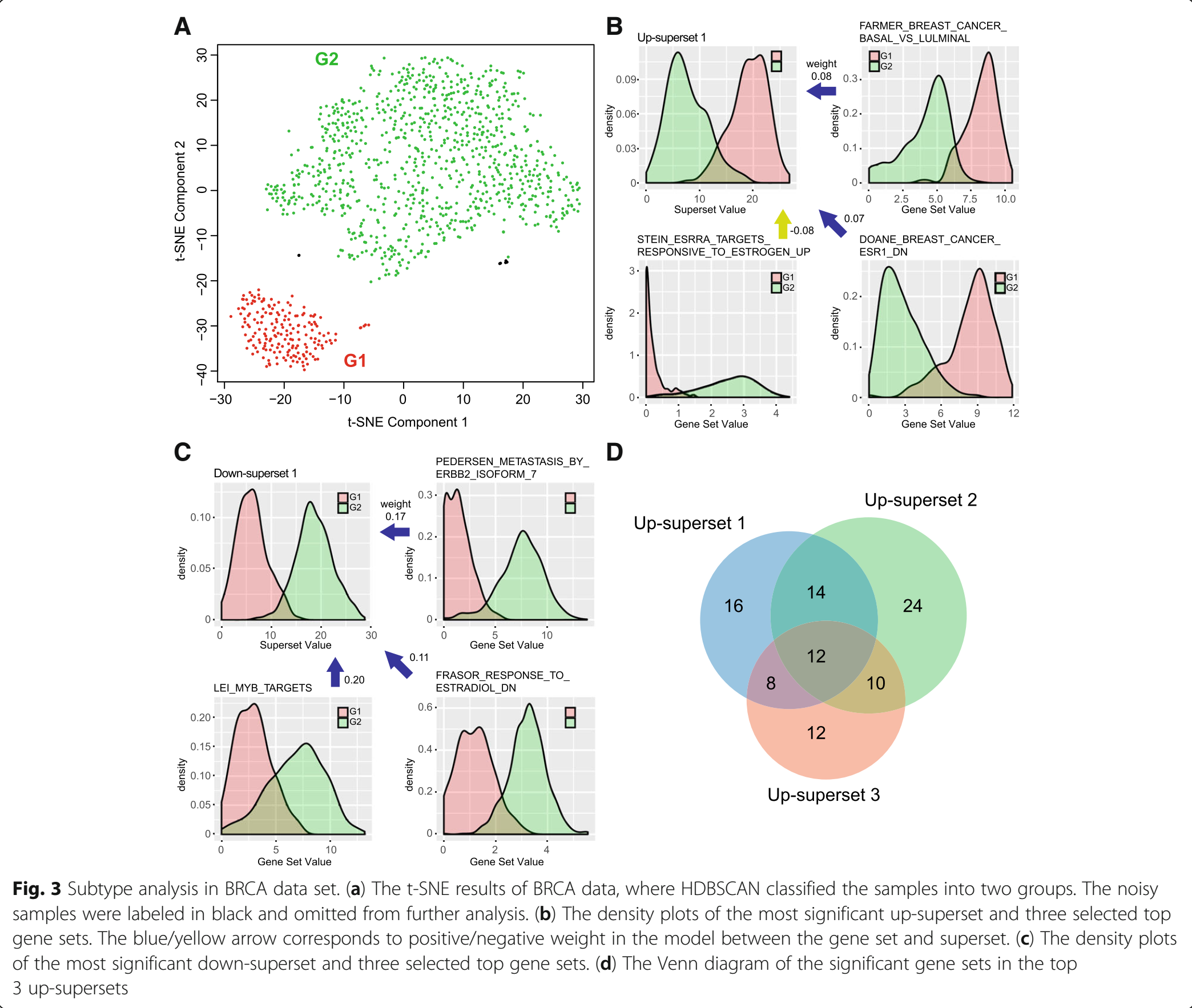
结果如上图所示。
- 依然使用\(\mu\gt1\)和\(\sigma\gt0.5\)的标准,得到15183 genes和1099 samples,然后进行t-SNE,使用HDBSCAN可以自然地将其聚类为两个亚型:G1(red)和G2(green)。
- 基于WWU test(location shift mu=9,\(P\lt0.01\)),可以确定4个up-supersets和3个down-supersets(Tab S1)。
- gsScore > 2 sd(gsScore在superset中的标准差)的gene sets被视为high impact gene sets。
最显著的up-superset和down-superset的top15 gene sets如下所示,其中的\(PScore=-\log_{10}(P-value)\):
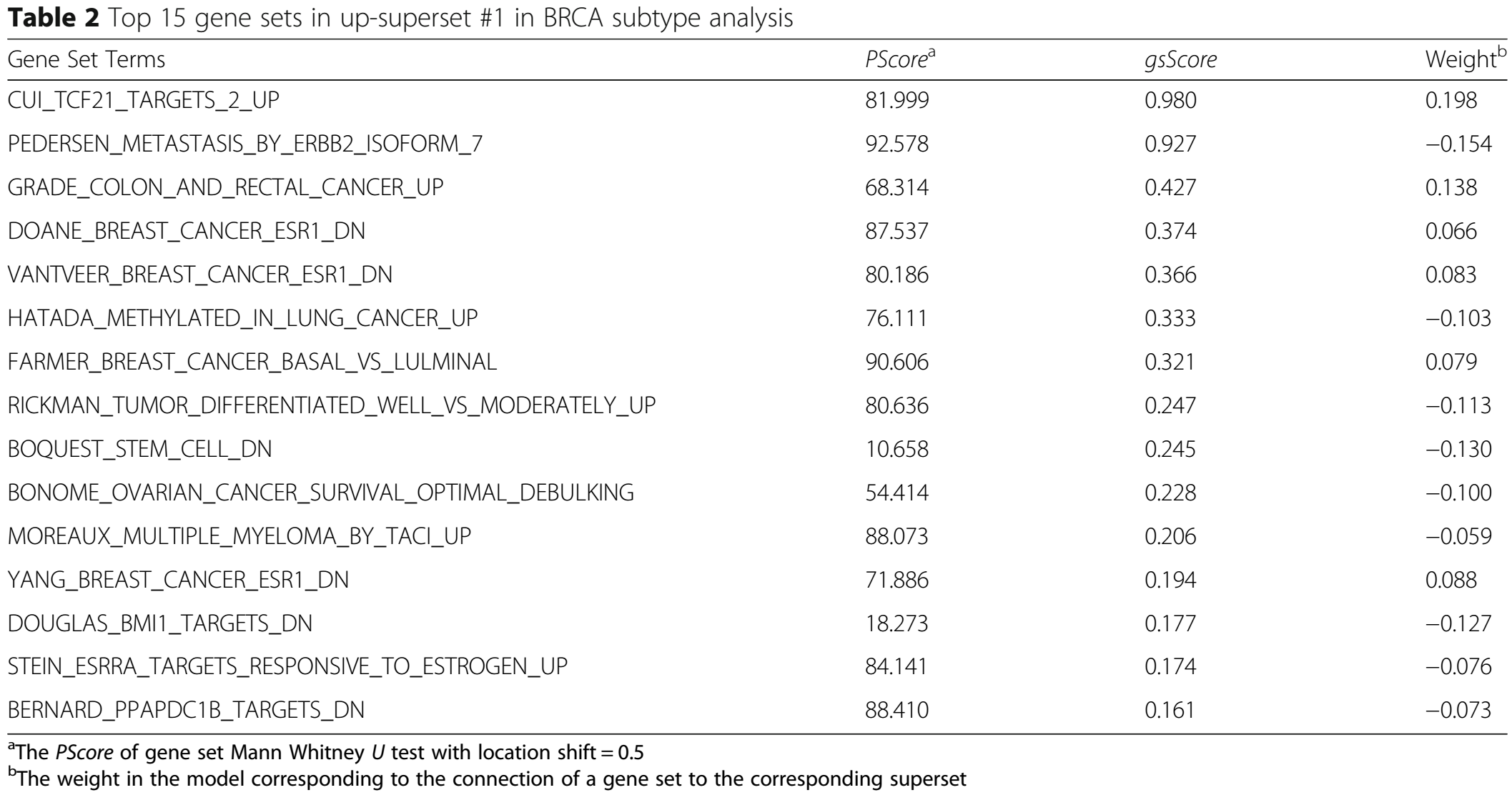
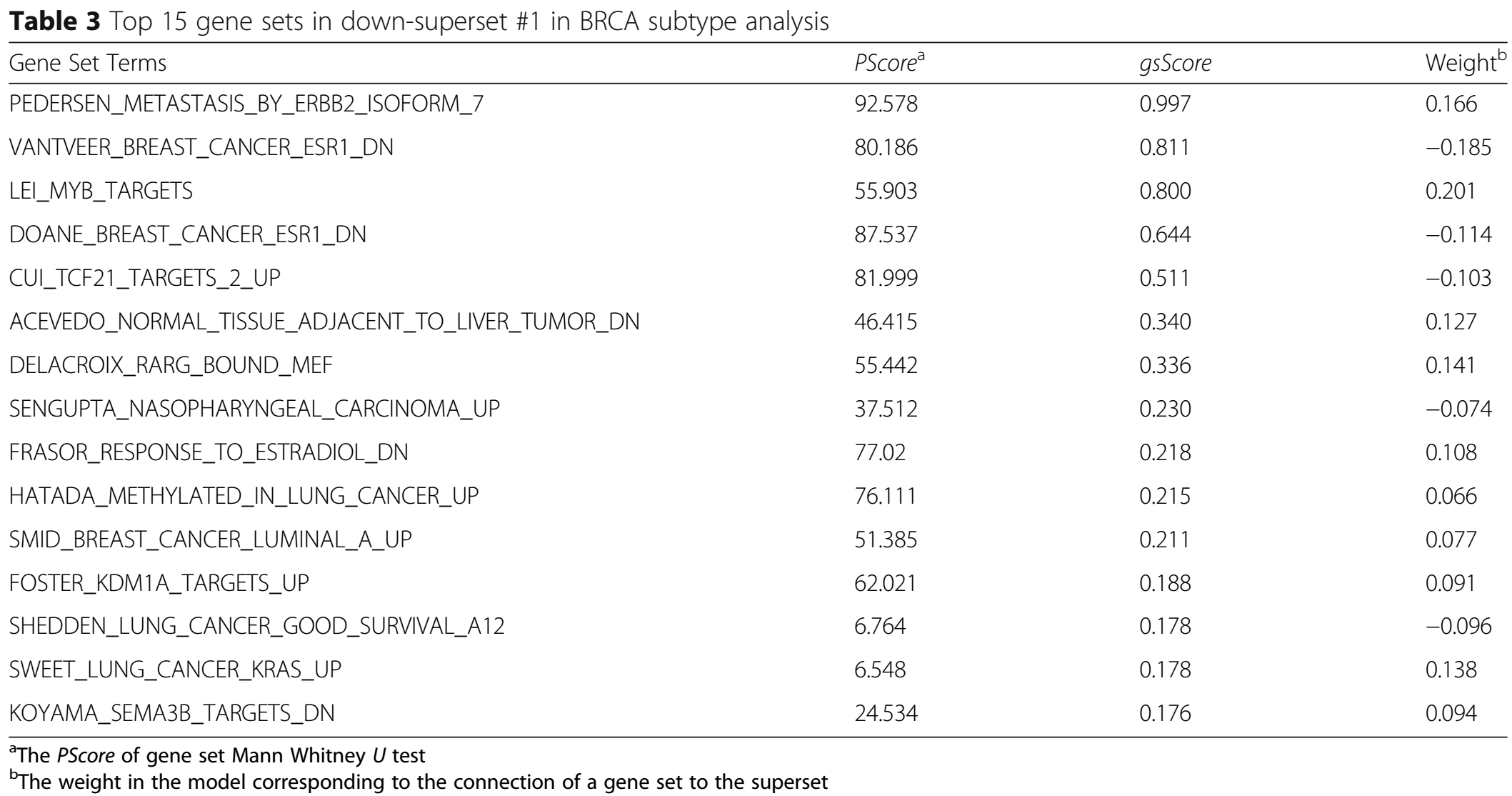
其中有许多与乳腺癌亚型高度相关的gene sets【24,25,26,27,28】。
之后,我们对G1/G2也进行了GSEA分析,发现124个high impact gene sets中的53个(42.7%)也是GSEA的富集gene sets(P<0.05),说明了GSAE方法找到的这些super sets是合理的。
另外,top3 up-supersets的Venn diagram展示在上面的fig 3d中,其中的大多数城府gene sets和Basal subtype相关,Up-superset 1另外相关于estrogen related gene sets,up-superset 2有许多gene sets相关于ERBB2。
使用superset分类器预测乳腺癌PAM50亚型
将decoder替换为了一个softmax分类器,则我们的模型可进行分类。
使用的数据集是821 x 15183的BRCA样本,对PAM50亚型(Basal、LumA、LumB和Her2)进行分类,使用10-CV进行验证,结果如下所示:
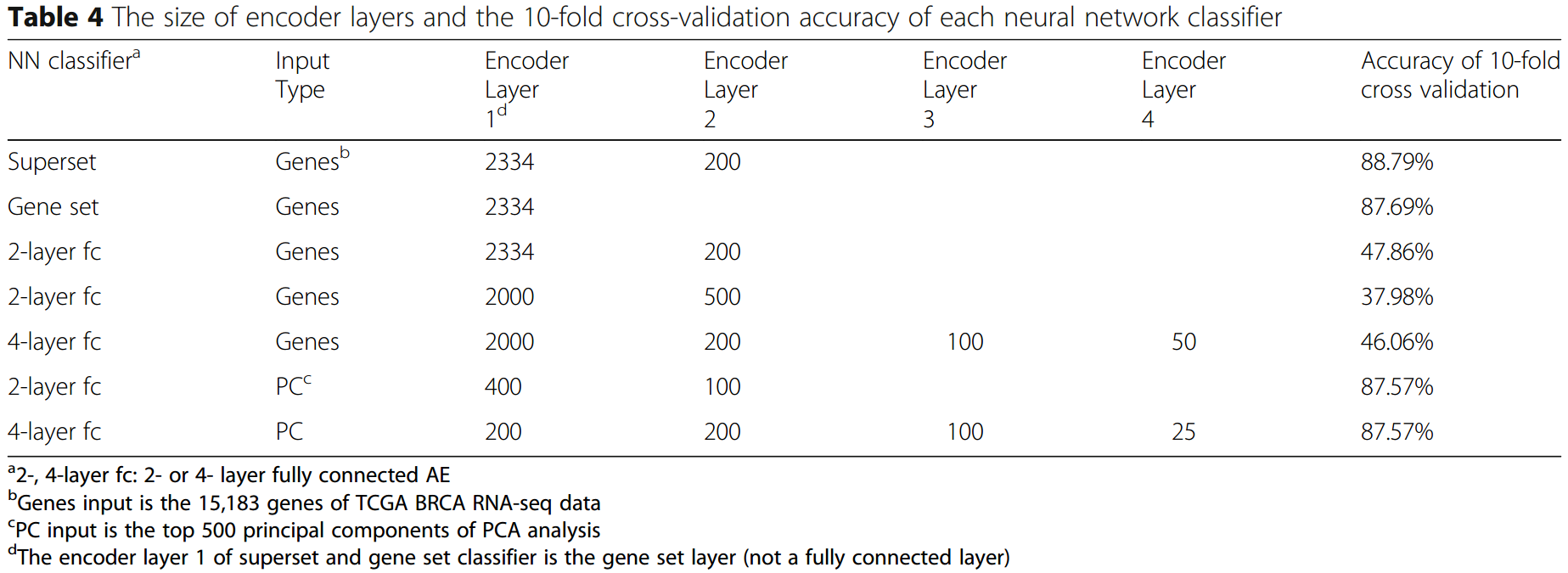
其中Gene set指的是没有superset layer,直接将gene sets layer的结果输入分类器;而n-fc表示使用n层的fc搭建的MLP作为encoder。可以看到,superset layer得到了最好的结果。
之后,通过10次10-CV,我们计算了分类器的灵敏度和特异度:
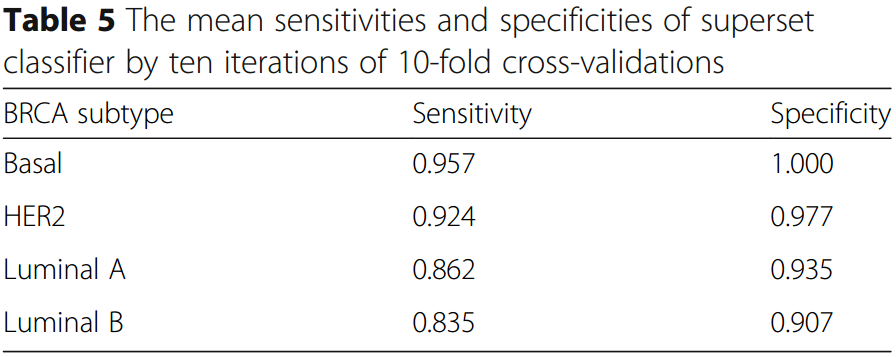
肺腺癌数据的生存分析
数据集:TCGA LUAD,515 x 15188。
首先使用log-rank test确定了6个supersets。然后通过gsScore选择出每个superset中的top20 gene sets(Additional File 3: Table S2),这些gene sets在gene set level上的log-rank test也是显著的。
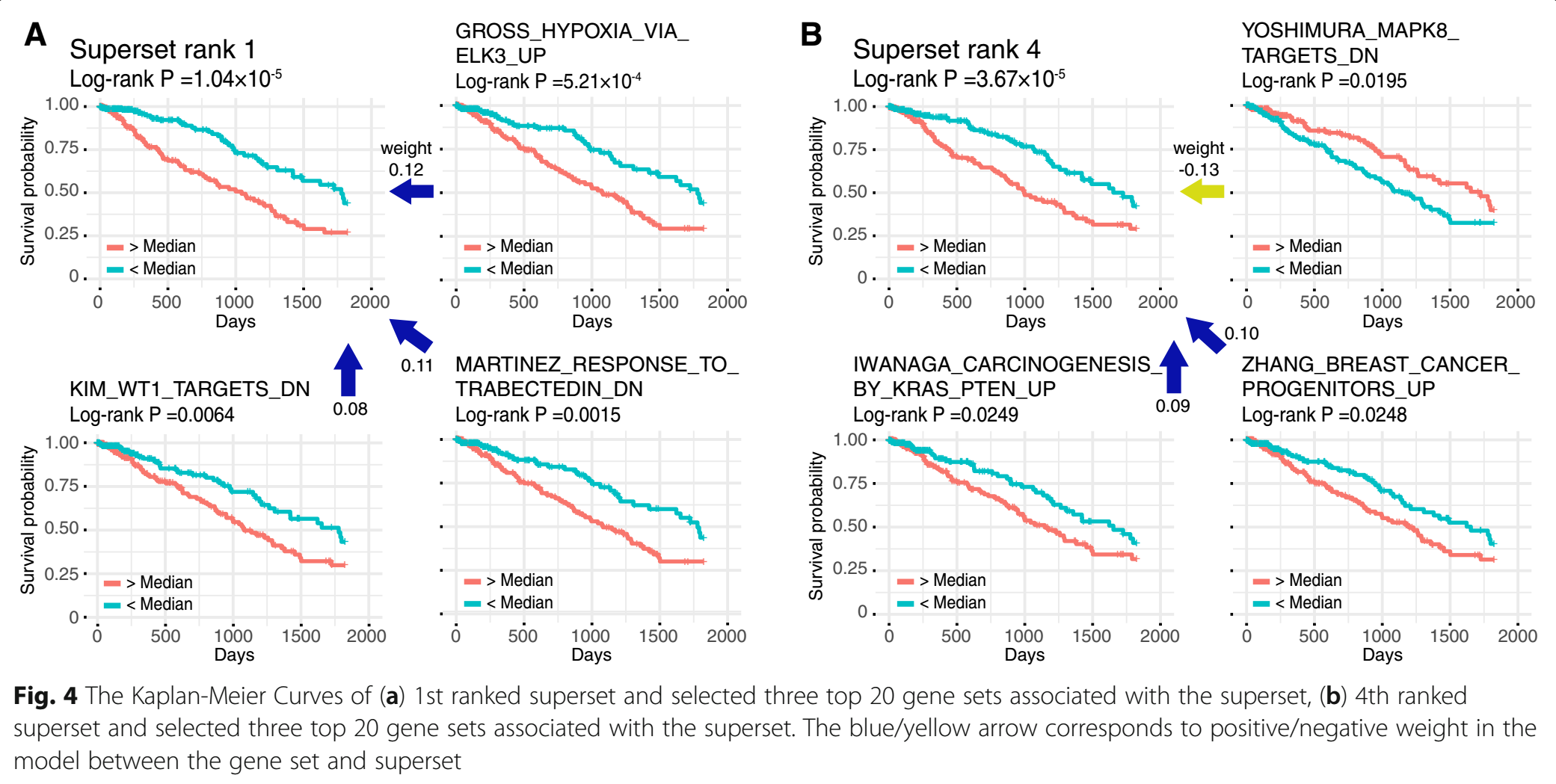
以下是选择的第1和第4顺位的supersets及其top15 genes(选择第4是因为其与第1的重叠gene sets是最少的):
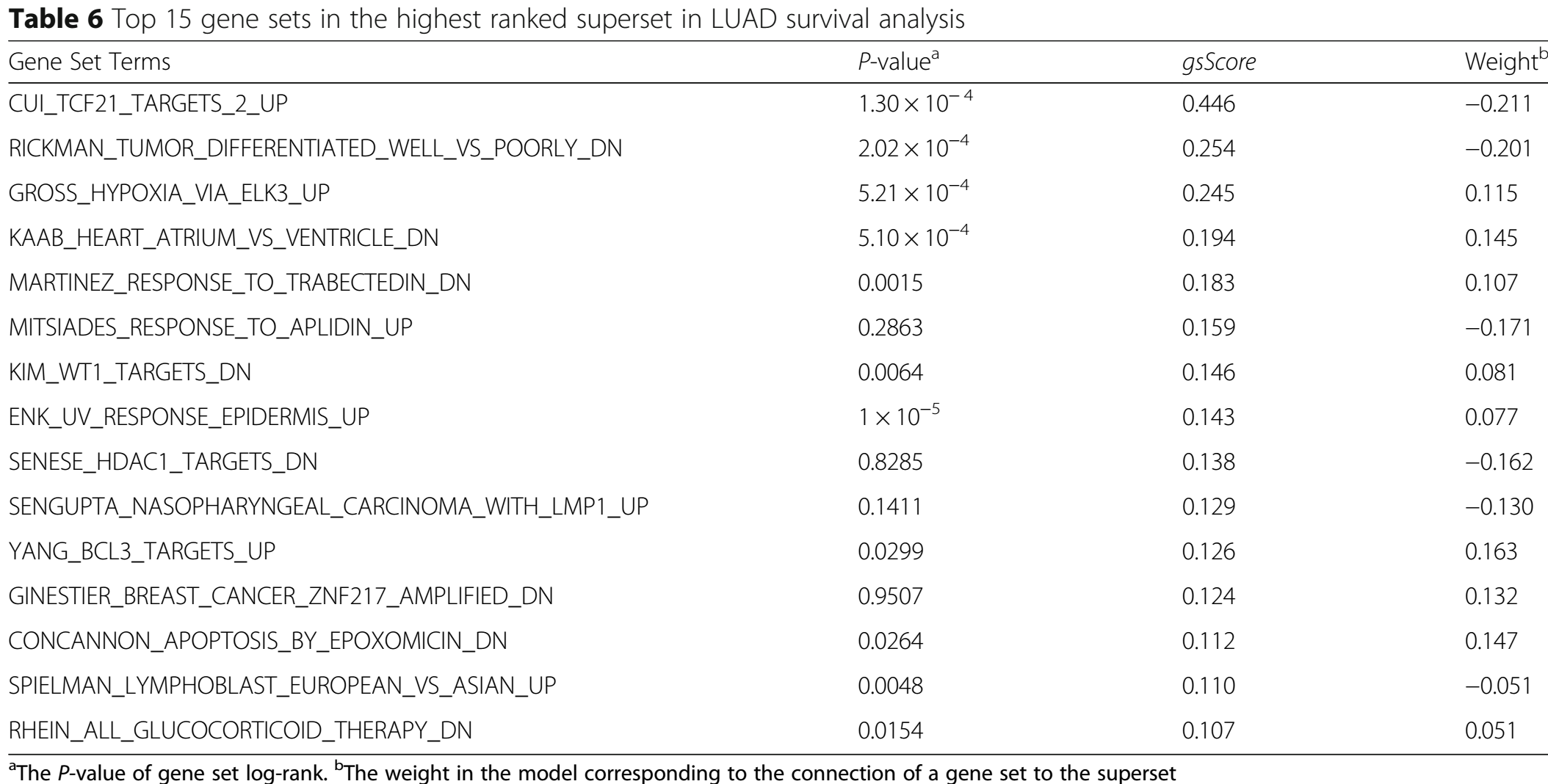
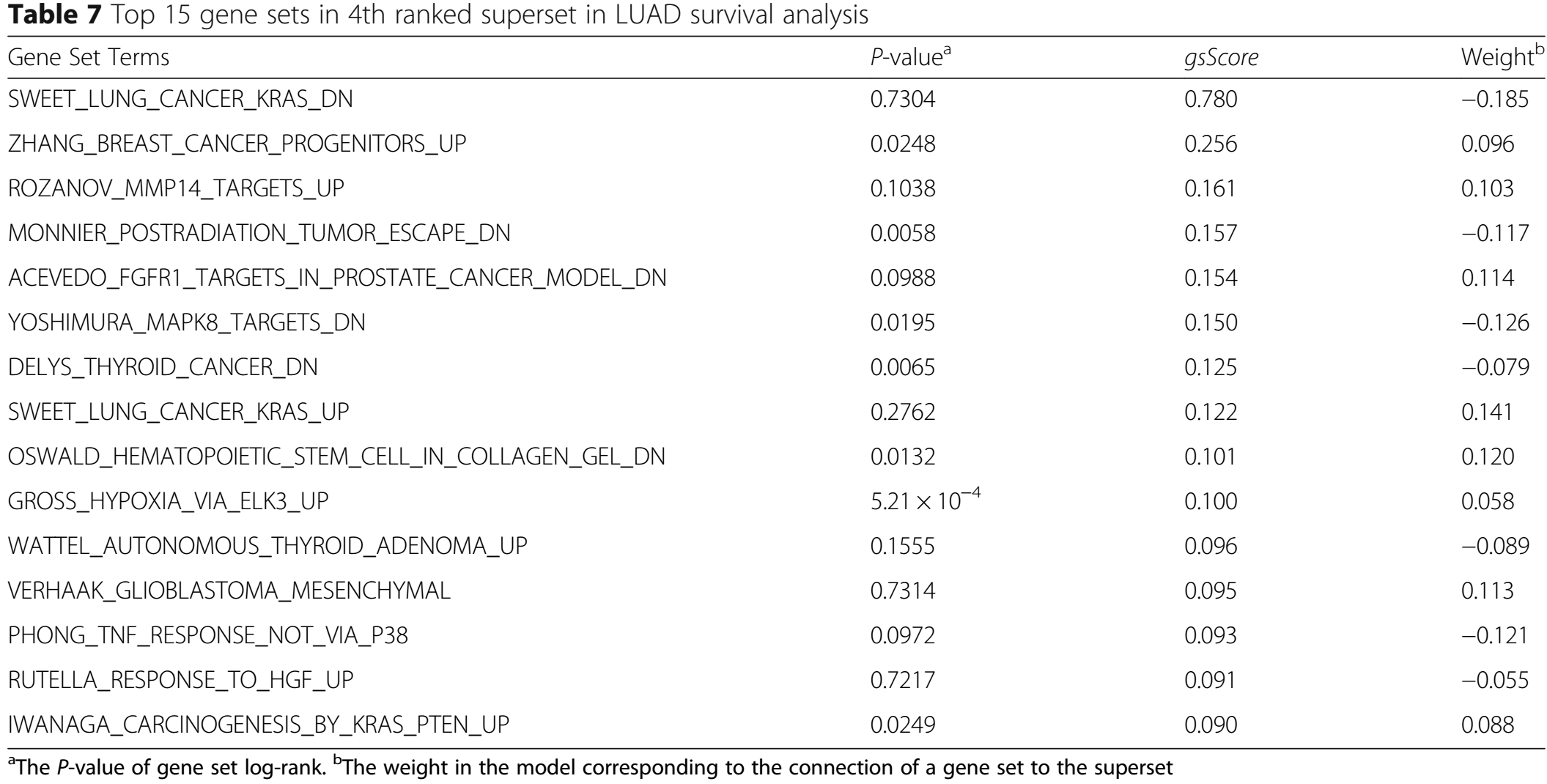
许多文献研究也支持了以上的结果【33-43】。
supersets可以提高生存分析的重现性
这里使用BRCA、LUAD、SKCM和LGG 4个数据来验证,使用\(\mu\gt1\)和\(\sigma\gt0.5\)来筛选变量,然后分割60%和40%作为train和test set。在train set上训练GSAE,在train和test set上得到superset表示。之后在train、test set上使用median分组,并进行log-rank检验来得到显著的生存相关supersets和gene sets。然后我们来比较train和test上得到的显著supersets、gene sets的相似性,使用的指标是Jaccard index和proportions z-test。结果如下所示:
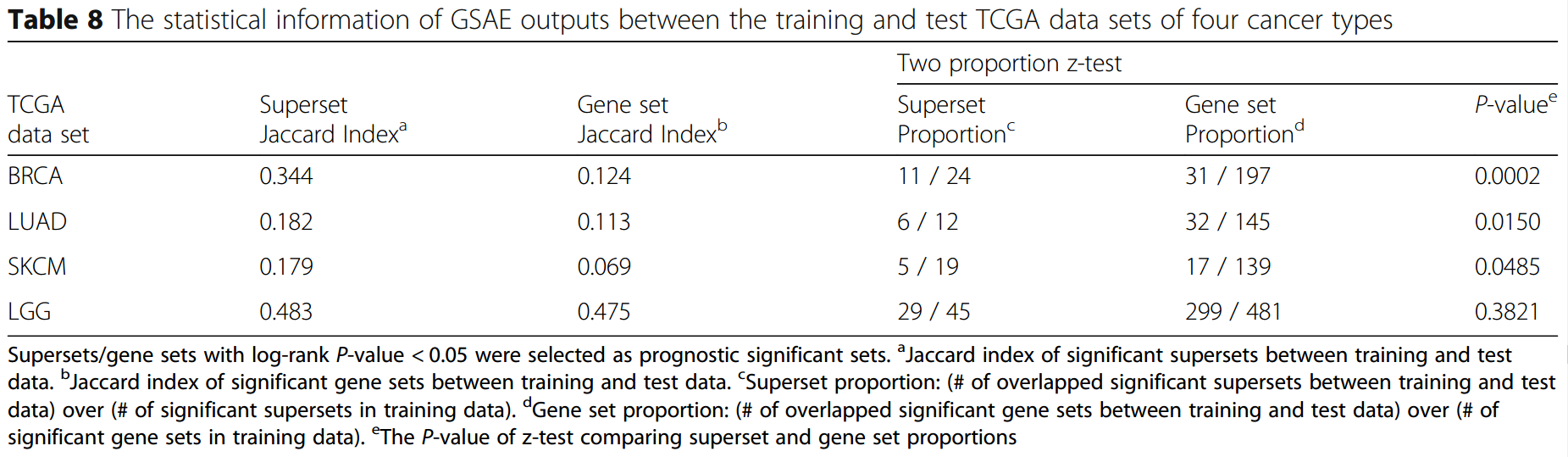
可以看到,总体来说,supersets的重现性要好于gene sets。
Discussion
和其他的机器学习方法类似,GSAE方法尽管在不同的超参数设置下可以得到类似的loss和分类结果,但其学习得到的supersets可能是不同的,那些更加显著的supersets可能有更高的频率出现。导致这个问题的原因可能来自gene sets的重叠和权重的初始化。
该算法的另一个问题在于需要大量的样本量。这可能意味着GSAE可能更加适合单细胞数据,single-cell RNAseq的测序深度低,但有更多的样本量。
supersets的概念不仅提供了更强的复现性,而且也为理解gene sets间的依赖性提供了一个机会。



