Convolutional neural network models for cancer type prediction based on gene expression
- 杂志: None
- IF: None
- 分区: None
- github
Introduction
癌症是人类的第二大死因,大量的研究旨在能够提高癌症诊断和治疗,癌种分类旨在通过现有的数据来学习一个分类器去完成自动化的癌症预测分类过程。
DL是人工智能的一个分支,配合如TCGA【7】这样的大规模测序数据,其得到了极大的发展:
- 【8】使用6703 tumor和6402 normal samples训练了一个MLP
- 【9】使用一个CNN model来进行所有33类癌症的分类,并得到了超过95%的准确率;【10】则利用KNN算法,并结合试图遗传算法进行gene筛选,达到了90%的准确率,在31类分类上
- 【12】将genes分为两类:oncogenes和tumor suppressors来确定癌症状态;transcription factors来将癌症分类到起始的组织。该研究在10-CV上得到了97.8%的ACC
- DeepCNA【13】是一个基于CNN的分类器,使用的是来自COSMICS【14】的copy number aberrations(CNAs)数据和来自两个人类细胞系的HiC数据,在25类癌症分类上达到了60%的效果
虽然以上算法都得到了非常好的精度,但其都没有或很少讨论如何去除组织差异所带来的影响。基于以上研究得到的biomarkers我们无法确定是tissue-specific genes还是cancer-type-specific genes。
还有“我们”在之前的一项研究【15】中提出了一个基于AEs的模型——GSAE,其可以将功能通路或功能基因集的信息进行提取并得到可解释的结果。“我们”成功应用GSAE进行乳腺癌亚型分类。
本研究试图来探索不同类型的CNNs model进行癌症分类的效果,系统的研究了不同容量的卷积核的效果。此外,本研究还提出了一种新的模型解释性方法来筛选关键基因。
Methods
数据集
- 使用TCGAbiolinks【17】 package下载pan-cancer RNA-seq数据,其中包括10340个33类癌症样本和713个正常组织样本。
- 得到\(\log_2(FPKM+1)\)。
- 在所有样本上,使用mean < 0.5和std < 0.8删除低信息量的genes,最终得到7091个genes。
- 另外还收集了964个BRCA样本的PAM50亚型信息,用于验证模型的鲁棒性。
- 为了验证模型的鲁棒性,我们为每个gene加入了高斯噪声(\(N(0,k\mu)\),其中\(\mu_i\)是第\(i\)个gene的平均表达量,\(k\in\mathcal{U}(0,5)\))。最后如果得到的gene的表达量小于0,将其置为0。
CNN模型设计
- 本研究不再研究gene排序所带来的影响,所有的gene将被排序在一个特定的顺序上。
- 本研究将着重研究CNN kernels的设计来学习genes间相关性的能力。
- 本研究中我们将只使用一层的CNN,shallow model可能在癌症分类这样的问题上已经是足够的,更深的模型在生物学数据上可能并不会带来性能的提示【18】,而且这也减轻了过拟合的风险【19,20】,提高了训练效率。
矢量化输入的CNN
接受的是一维向量,使用1-D CNN,之后进行一次maxpooling,然后接一个FC layer和softmax layer进行预测。
因为这里我们并不确定相邻的gene间是否存在相关性,所以这里将stride设置的和kernel size一样大,从而提取一个kernel的整体信息。
矩阵输入的CNN
类似【9】,将样本reshape成2-D matrix,使用2-D CNN提取信息。同样是CNN-maxpooing-fc-softmax的架构。这称为2D-Vanilla-CNN。
矩阵输入的CNN,但使用1D kernel
这里使用两个1-D CNN(一个vertically、一个horizontally)来对matrix输入提取特征,之后再将两个的结果concat进入fc layer,可见后面的fig1c。
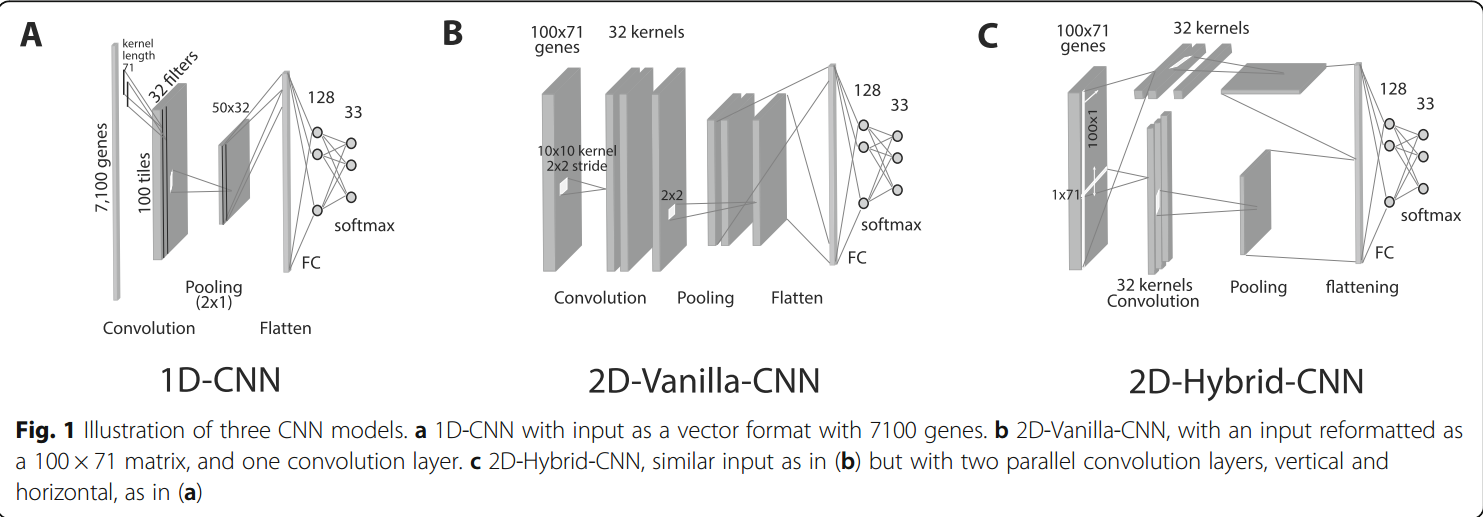
另外,“我们”还实现了【9】中的model,称为2D-3layer-CNN,以便进行比较。
CNN模型解释
利用keras visualization package——kears-vis【22】的guided gradient saliency visualization方法,得到每个样本在每个gene、每类癌症上的重要性评分,然后在样本上进行平均,得到一个gene-effect matrix(\(7091\times 34\)),然后将值归一化到\([0,1]\)中。大于0.5的值被认为该gene是一个marker。
Results
模型设置和超参数
使用Keras实现。
1D-CNN的输入是gene按照其字母顺序排序得到的1D vector。
2D输入是以上vector被reshape成100 x 71的matrix。
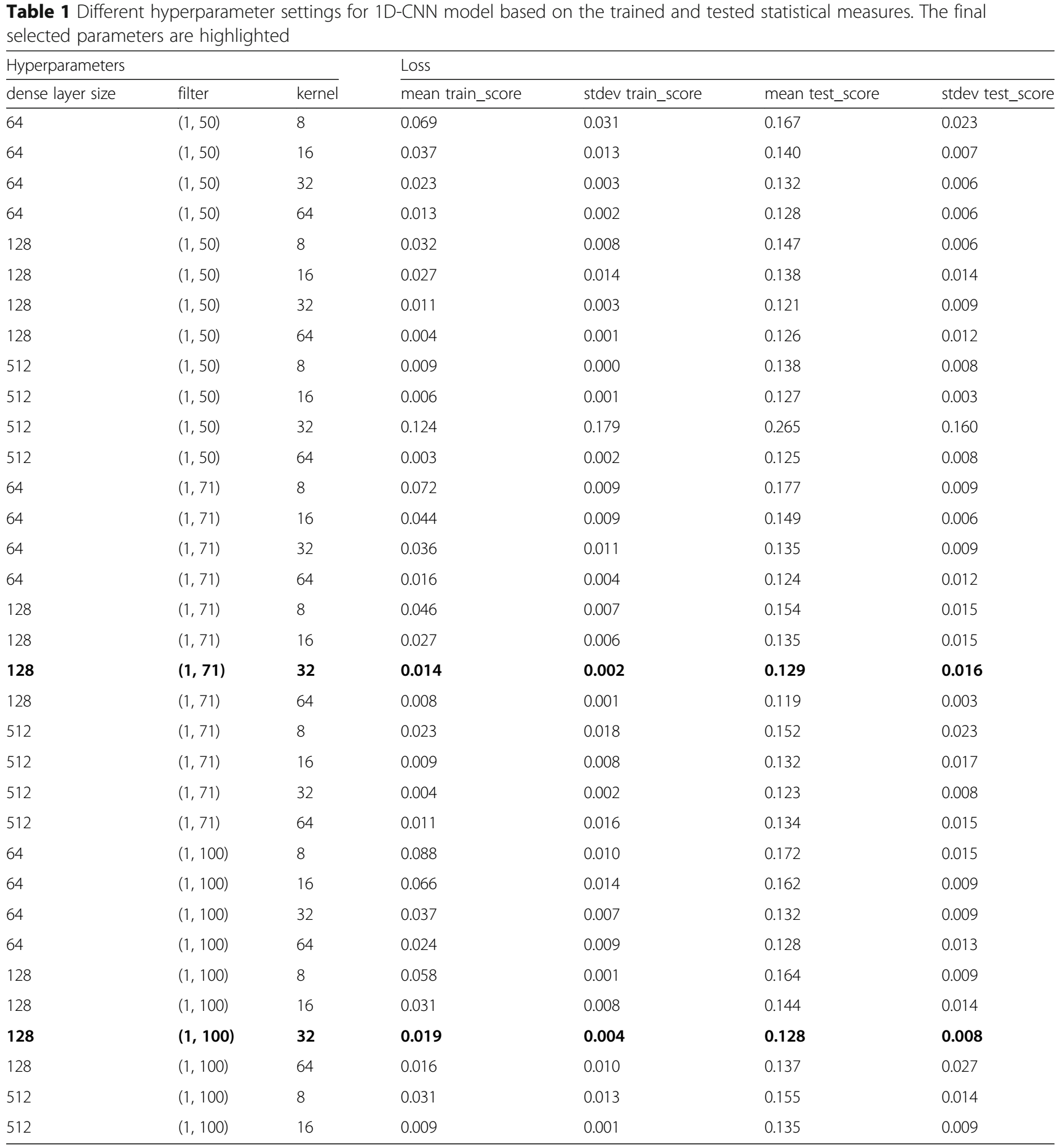
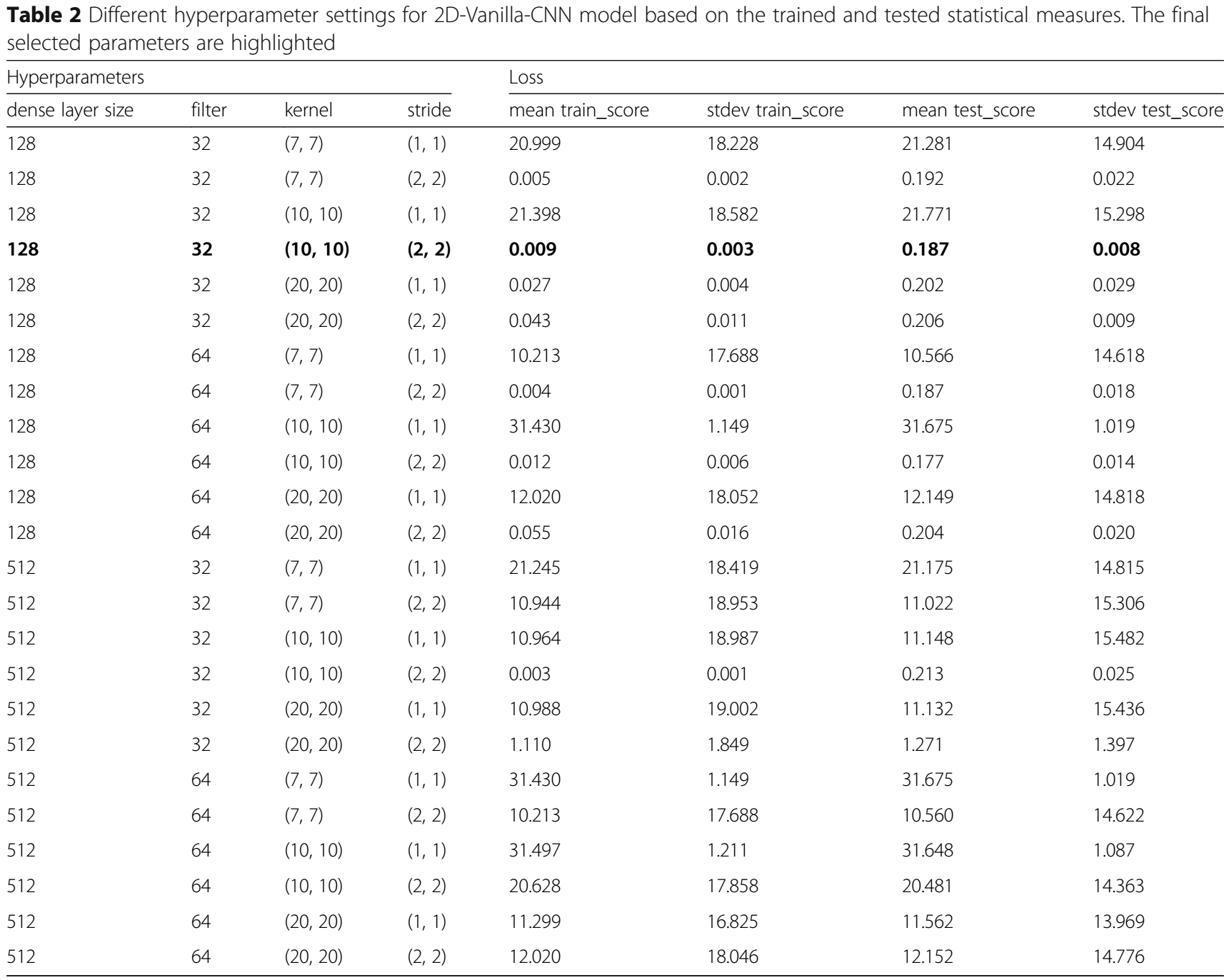
4个最重要的超参数:kernel的数量和大小、stride、fc layer的nodes数量,展示在下面的table 1和2中,使用【25】中提供的Grid search方法进行挑选。
loss是cross entropy,Adam optimizer,epoch和batch size是50和128,early stop的patience是4,激活函数是ReLU。
进行训练时没有validation,early stop是利用的train进行的(提前使用8/2的train-validation分割来验证了,在10个epochs后基本就收敛了,而且没有过拟合,这个在fig2a中)。
可以看到【9】的模型的收敛是最慢的
将5-CV重复了6次,计算acc的均值和方差。
结果在fig2b中,1D-CNN、2D-Vanilla-CNN和2D-Hybrid-CNN分别是\(95.5\pm0.1\%\),\(94.87\pm0.04\%\),\(95.7\pm0.1\%\)。
这里的score是什么?
组织特异性的影响
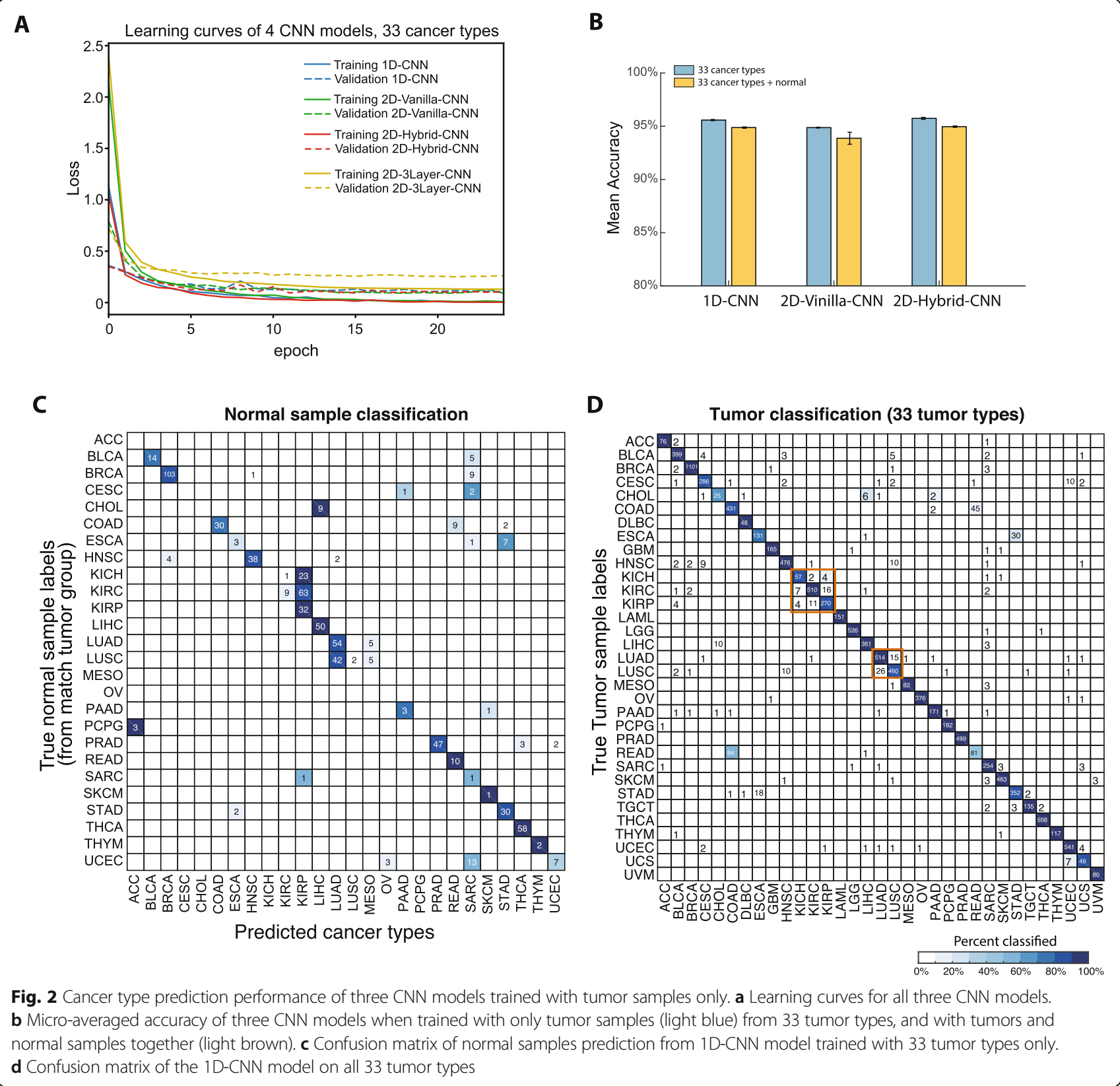
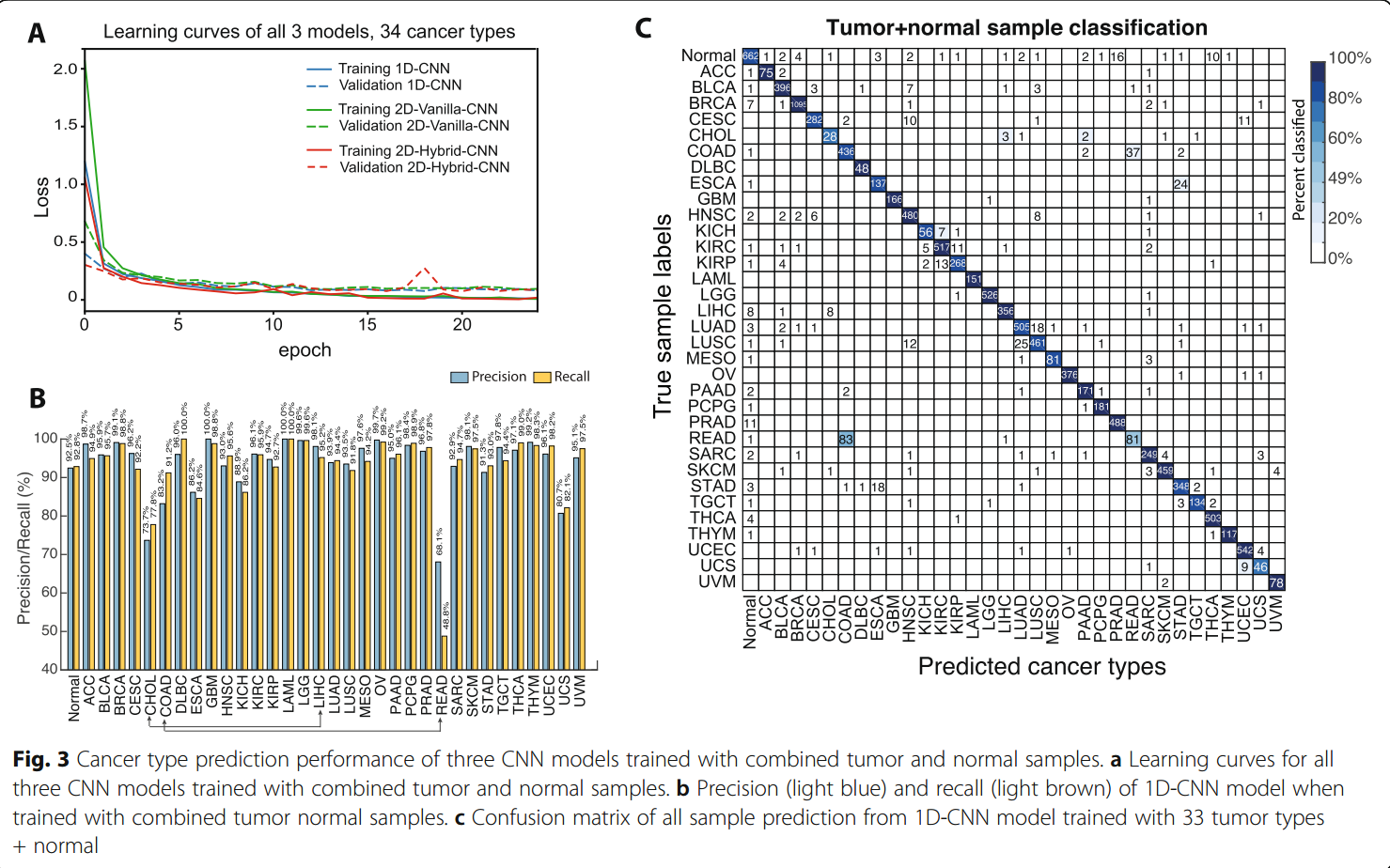
我们首先训练了两个模型:只使用tumor样本和只使用normal样本的1D-CNN。fig2中的混淆矩阵显示,两个模型所出现的错误分类显示出相似的组织特异性,即在解剖学上位置相近的组织分类直接更容易相互分错类,这意味着模型学习到组织特异性而不是癌症特异性的风险很大。
为了克服这个问题,策略是将normal的样本也加入其中,作为新的一类。结果显示(fig2b),预测acc相比33类分类,只有轻微的下降(1D-CNN、2D-Vanilla-CNN和2D-Hybrid-CNN分别是\(94.5\pm0.1\%\),\(93.9\pm0.6\%\),\(95.0\pm0.1\%\))。
进一步,分析了1D-CNN的micro-averaged precision-recall statistics(fig3b)。可以看到其中READ、CHOL、COAD等效果有比较大的差距。
探索cancer marker genes
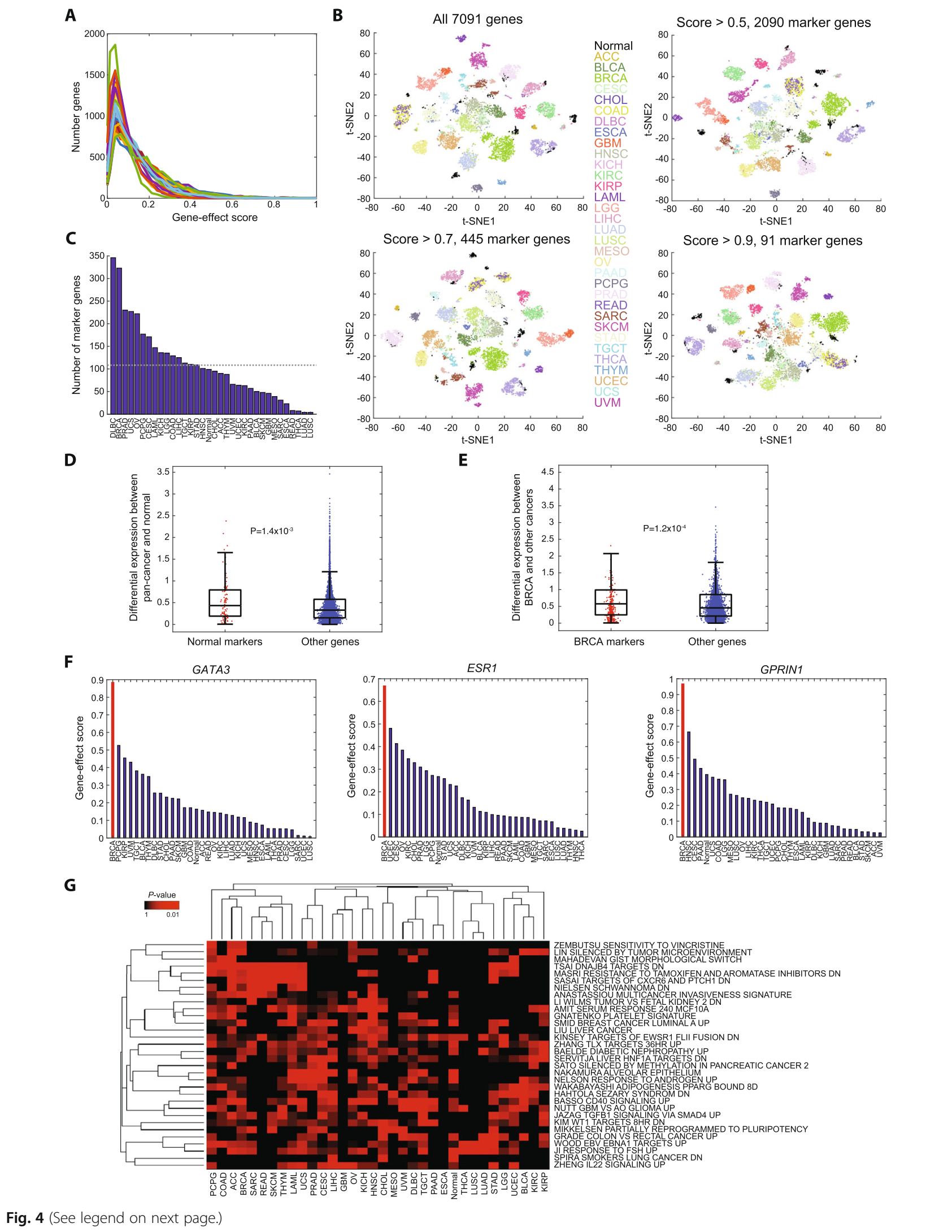
- 探索了所有cancer types的saliency maps的gene-effect scores的分布,发现都服从power law(fig4a)。
- 凭借gene-effect scores逐步减少gene数量(0.5 -- 2090 genes,0.9 -- 91 genes),然后使用tSNE可视化(fig4b)。
- 之后使用scores > 0.5进行后续分析,一共得到3683个markers(不重复的有2090个),平均每个癌症类型有108个(fig4c)。进一步比较每类得到的markers数量和分类精度,可知,在分类精度越小的癌症类别上,其筛选得到的变量越少。
进一步,“我们”比较了normal markers(99)和BRCA markers(323)在normal vs others和BRCA vs others上的表达差异分布,发现确实存在差异(fig4d-e)。
在BRCA markers中,确实出现了诸如GATA3【26】和ESR1【27】等众所周知的makers,也发现了一些新的markers(GPRIN1、EFNB1、FABP4)(fig4f)。
接下来,“我们”对每一类得到的markers进行功能富集分析(MSigDB【28,29】),one-tailed Fisher's exact test(P < 0.001)得到了32个相关功能,其中确实有许多的癌症相关功能(fig4g)。

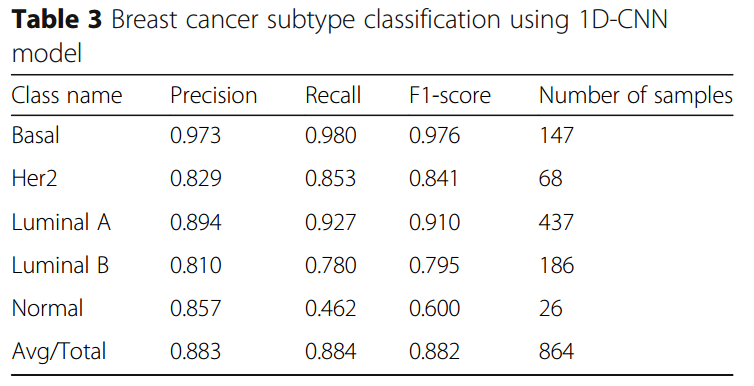
乳腺癌亚型预测
预测4个亚型+normal样本,得到了88.3%的ACC。
Discussion
我们进一步总结了这些模型的参数量和表现等等:
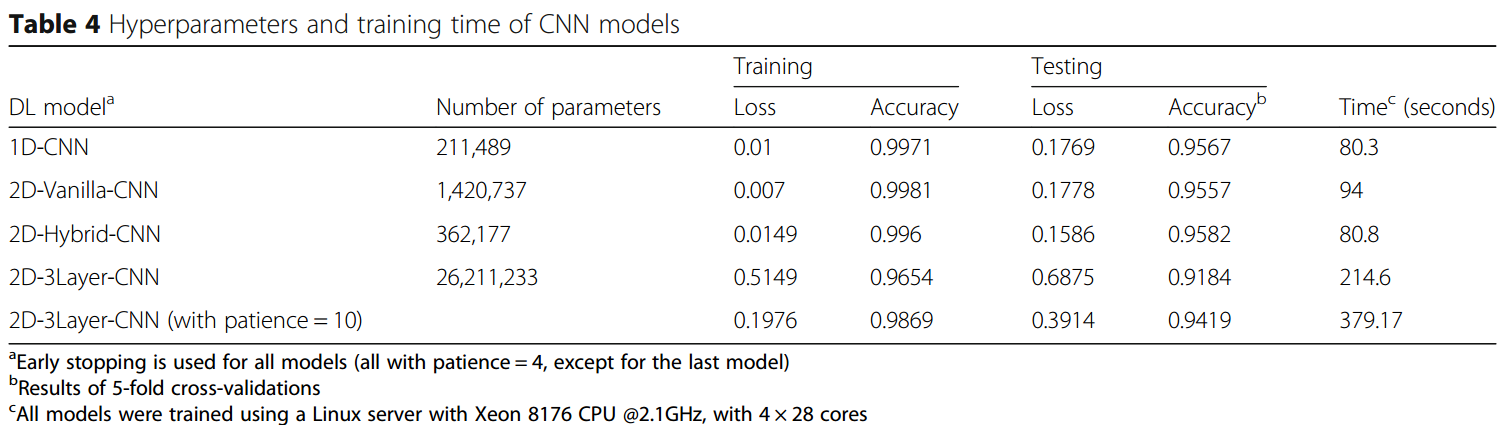
可以看到,在基因组学上的模型,可能并不需要太过复杂,仅仅一层的1D-CNN就可以达到不错的效果,而且可能因为参数量的较少而使得其过拟合的风险大大降低。2D-Hybrid-CNN因为可以从两个方向来提取信息,所以其效果要略好一些。
2D-Vanilla-CNN虽然效果相似,但其参数量提高了5倍还多,而且有更多的需要调节的超参数。“我们”通过为输入添加不同比例的噪声来比较1D-CNN和2D-Vanillia-CNN的鲁棒性,结果如下:
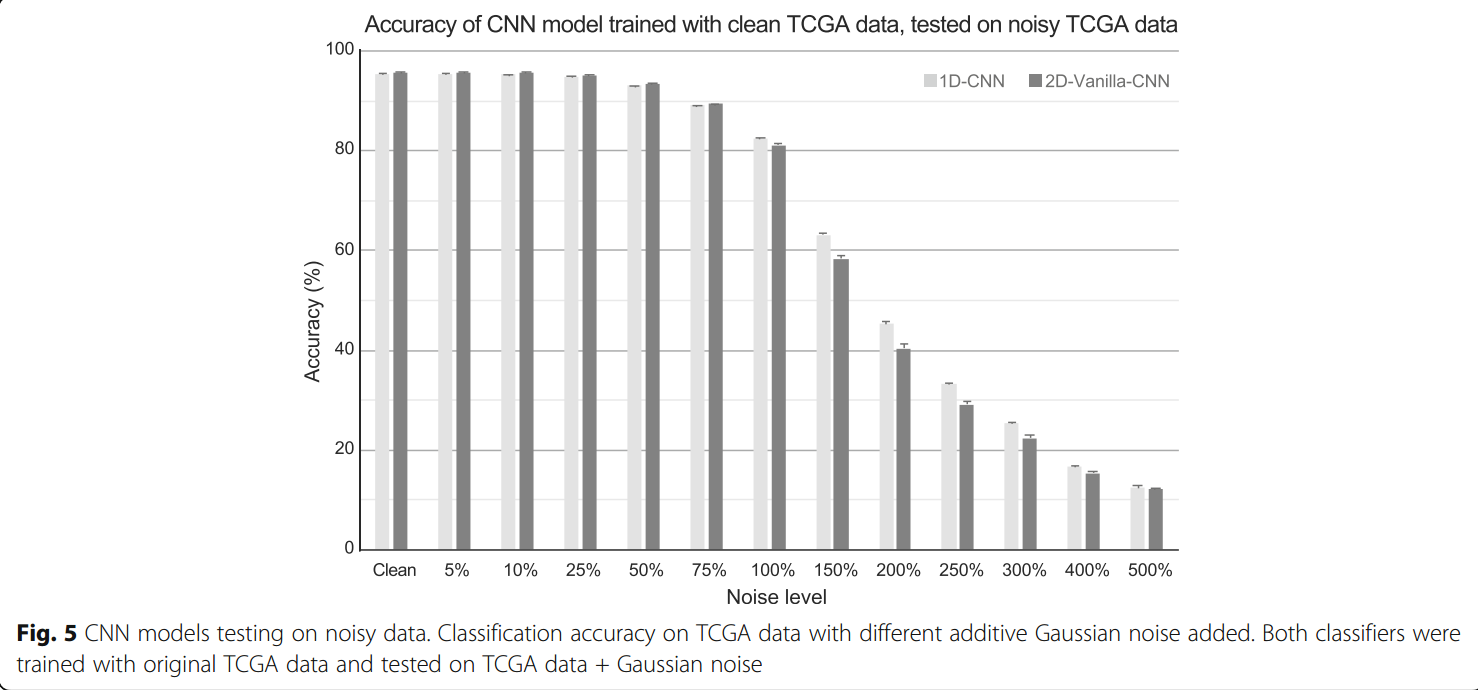
尽管两者的行为非常相似,但1D-CNN的鲁棒性还是要好一些。
未来的研究方向:
- 将GTEx中的转录数据利用【33】
- 利用到其他基因组学数据,如DNA mutation、copy number variation和DNA methylation等。



